 2018, Vol. 35
2018, Vol. 35文章信息
- 邓丽颖, 杨灿宇, 闫亚辉, 李佳伟, 岑如月, 赵辉
- DENG Liying, YANG Canyu, YAN Yahui, LI Jiawei, CEN Ruyue, ZHAO Hui
- 桦褐孔菌三萜的酰化对其抗肿瘤活性的影响
- Synthesis of acylated triterpenes of Inonotus obliquus and their antitumor activities
- 天津中医药, 2018, 35(11): 868-873
- Tianjin Journal of Traditional Chinese Medicine, 2018, 35(11): 868-873
- http://dx.doi.org/10.11656/j.issn.1672-1519.2018.11.18
-
文章历史
- 收稿日期: 2018-05-30

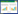



2. 河南大学药学院, 开封 475004
桦褐孔菌[Inonotus obliquus(Fr.)Pilat]为担子菌亚门、多孔菌目、刺革菌科的药用真菌,在亚洲、欧洲及北美洲均有分布,它作为抗肿瘤药物在民间使用已有很长历史[1-2]。现代药理学实验证明桦褐孔菌可抑制NCI-H460人大细胞肺癌细胞、HT29人结肠癌细胞及B16-F10小鼠黑色素瘤细胞等多种肿瘤细胞的生长,并可逆转Hep2/R肺癌细胞的多药耐药性[3-4]。桦褐孔菌的化学成分主要为羊毛脂烷型三萜类化合物[5-7],其中以inotodiol和trametenolic acid 2个化合物的含量最高,他们也是桦褐孔菌中众多其他次生代谢产物的生物原料。本实验拟从桦褐孔菌中分离制备inotodiol、trametenolic acid及抗肿瘤活性化合物inonotusane A,并以此为原料制备其酰化产物,进而考察其结构变化前后的抗肿瘤活性,为桦褐孔菌三萜的结构修饰方向提供参考,inotodiol、trametenolic acid及inonotusane A的结构见图 1。

|
| 图 1 目标化合物1(a-c)、2a和3a的合成路线 Fig. 1 Systhesis route of the objective compounds 1(a-c), 2a and 3a |
AVANCE400M核磁共振仪(1H HNMR: 400 MHz,13C NMR: 100 MHz,德国Bruker公司);高分辨质谱(安捷伦公司);VERTEX70傅立叶变换红外光谱仪(Bruker公司);XT6显微熔点测定仪(北京市科技电光仪器厂);制备型高效液相色谱(HPLC)仪(日本岛津),制备型HPLC色谱柱为Shim-pack PREP-ODS(H)KIT column(250 mm × 20 mm, 5 μm);十八烷基键合固定相(Rp-18)柱层析材料(Merck公司),Rp-18高效薄层预制板(Merck公司),柱色谱用硅胶200~300目(青岛海洋化工厂);色谱甲醇(天津市四友精细化学品有限公司),本实验所用其他化学试剂均为分析纯。
本实验所用药材采自吉林省珲春地区,由本实验室鉴定为Inonotus obliquus(Fr.)Pil。紫杉醇注射液(5 mL, 30 mg)购自海南中化联合制药工业股份有限公司,批号20130806。
2 实验方法 2.1 原料化合物的分离与结构鉴定取干燥粉碎的桦褐孔菌子实体10 kg,采用95%乙醇回流提取5次,每次1 h,得到乙醇提取物430.5 g。将乙醇提取物均匀分散于水中,依次用石油醚、乙酸乙酯、正丁醇萃取(15 L×3)得到各萃取物。将乙酸乙酯萃取物(198.2 g)用硅胶柱层析(石油醚-乙酸乙酯)粗分为Fr. 1~10共10部分。将馏分Fr. 2再用硅胶柱层析(石油醚-乙酸乙酯μ从100:0到0:100比例梯度洗脱)分离得到inotodiol(1);将馏分Fr. 3也用硅胶柱层析(石油醚-乙酸乙酯)分为3部分Fr. 3.1~3.3,进而将Fr. 3.1进行ODS开放柱层析(甲醇-水)和制备型HPLC(甲醇:水为77:23)进行分离,得到inonotusane A(3);将馏分Fr. 4采用ODS开放柱层析分为Fr. 4.1~4.5,再将Fr. 4.3采用硅胶柱层析(氯仿:甲醇:水为13:0.95:0.05)分离,得到trametenolic acid(2)。
2.2 目标化合物的制备化合物1(a-c)的制备:称取干燥的inotodiol(1)220 mg置于100 mL的圆底烧瓶中,加入约22 mL氯仿将化合物inotodiol溶解,另取506 mg的干燥琥珀酸酐,使用约8.8 mL N,N-二甲基甲酰胺(DMF)溶解后,加入圆底烧瓶中,再加入吡啶10 μL和4-二甲氨基吡啶(DMAP)适量,搅拌下加热至回流约7 h,TLC监测反应物反应完全。用旋转蒸发仪蒸出氯仿后,加入蒸馏水30 mL,用稀HCl调pH 2~3,再用乙酸乙酯(4×20 mL)萃取,合并乙酸乙酯萃取液,用70 mL饱和盐水洗涤4次,再加入无水硫酸钠干燥。次日,将有机相蒸干,硅胶柱层析(氯仿:甲醇:水为20:0.95:0.05)分离纯化后,用制备型HPLC分离,分别得到化合物1a(甲醇:水为77:23, tR 164.8 min)、1b(甲醇:水为77:23, tR 65.5 min)和1c(甲醇:水为84:16, tR 30.5 min)。
化合物2a的制备:称取干燥的trametenolic acid(2)320 mg置于100 mL的圆底烧瓶中,加入约10 mL吡啶将化合物trametenolic acid溶解,另取560 mg的干燥琥珀酸酐,用约10 mL DMF溶解后,加入圆底烧瓶中,再加入DMAP适量,搅拌下加热至83 ℃,反应约17 h,TLC监测反应物反应完全。将反应液用旋转蒸发仪旋蒸1 h后,加入蒸馏水20 mL,用稀HCl调pH 2~3,再用乙酸乙酯(4×20 mL)萃取,合并乙酸乙酯萃取液,用70 mL饱和盐水洗涤4次,再加入无水硫酸钠干燥。次日,将有机相蒸干,ODS开放柱层析(甲醇:水为90:10)分离纯化后,用硅胶柱层析(氯仿:甲醇:水为9:0.95:0.05)分离,得到化合物2a。
化合物3a的制备:称取干燥的inonotusane A(3)12 mg置于25 mL的圆底烧瓶中,加入约3 mL氯仿将化合物inonotusane A溶解,另取25 mg的干燥琥珀酸酐,用约1 mL DMF溶解后,加入圆底烧瓶中,再加入吡啶10 μL和DMAP适量,搅拌下加热至回流约10 h,TLC监测反应物反应完全。用旋转蒸发仪蒸出氯仿后,加入蒸馏水5 mL,用稀HCl调pH 2~3,再用乙酸乙酯(4×5 mL)萃取,合并乙酸乙酯萃取液,用20 mL饱和盐水洗涤4次,再加入无水硫酸钠干燥。次日,将有机相蒸干,制备型HPLC(甲醇:水为95:5)分离纯化后,用硅胶柱层析(氯仿:甲醇:水为8:0.95:0.05)分离,得到化合物3a。
2.3 体外抗肿瘤活性采用噻唑蓝(MTT)法测试化合物1-3及1a、1b、1c、2a、3a对A549人肺癌、Hela人宫颈癌、MCF-7人乳腺癌及4T1小鼠乳腺癌细胞株的抑制活性。实验中,受试化合物均用二甲基亚砜(DMSO)溶解,配制成10 mmol/L的母液,-20 ℃储存,临用前于37 ℃恒温解冻后,采用不加血清的培养基将母液稀释至不同浓度;对照品采用紫杉醇注射液,其中紫杉醇的浓度为7 mmol/L,将其按药品说明书进行储存,临用前采用不加血清的培养基将其稀释至不同浓度。
取对数生长期细胞,制备细胞悬液后,将细胞悬液均匀接种于96孔板中,每孔加入100 μL,将细胞放置于37 ℃、5% CO2的细胞培养箱中继续培养18~24 h;待细胞贴壁后,将培养基弃去,加入充分混匀的含有不同浓度药物(待测化合物及阳性对照化合物)的培养基至96孔板中,每孔加入100 μL,药物浓度分别为1、5、10、30、50 μmol/L,每种浓度设4个复孔,其中,调零组不接种细胞,空白组接种细胞但不加药;将细胞放置于37 ℃、5% CO2的细胞培养箱中继续培养48 h;将培养基吸出,每孔加入MTT试剂100 μL,将细胞放置于37 ℃、5% CO2的细胞培养箱中继续培养4 h;吸出上清液,每孔加DMSO 100 μL,振荡5~10 min,在酶联免疫检测仪490 nm处测量各孔的吸光度值,计算肿瘤细胞的抑制率及化合物的IC50值。
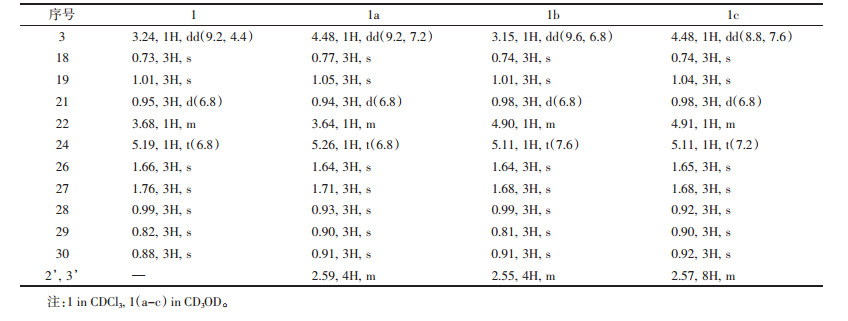
3 实验结果化合物1:白色粉末(1.2 g),ESI-MS m/z:465.57 [M+Na]+, 481.43 [M+K]+,其1H和13C NMR数据分别见表 1和表 2。通过与文献报道对照,将其鉴定为inotodiol[8]。
|
|
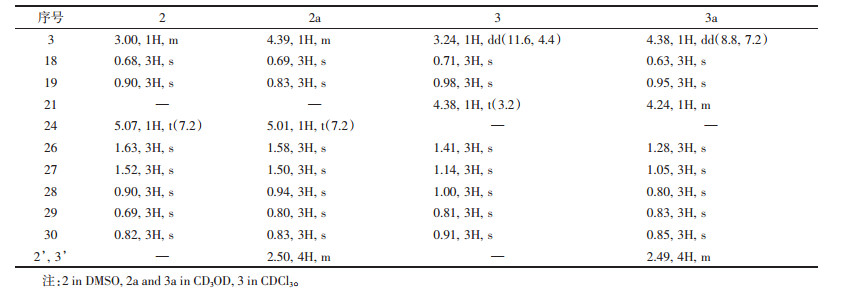
化合物2:白色粉末(802 mg),ESI-MS m/z:455.62 [M-H]-,其1H和13C NMR数据分别见表 2和表 3。通过与文献报道对照,将其鉴定为trametenolic acid[9]。
|
化合物3:白色粉末(23 mg, tR 43.7 min),HRESIMS确定其分子式为C30H50O3([M+Na]+, m/z 481.365 4; calcd 481.365 8),其1H和13C NMR数据分别见表 2和表 3。通过与文献报道对照,将其鉴定为inonotusane A[10]。
化合物1a:白色粉末(20 mg),回收率7.4%,HRESIMS确定其分子式为C34H54O5([M+Na]+, m/z 565.386 8; calcd 565.386 9)。IR(KBr)νmax: 3 450, 2 924, 1 727, 1 454, 1 375, 1 166 cm-1。1H NMR(400 MHz,CD3OD)数据见表 1,经与原料化合物1对照,并参考文献[11],3-H的化学位移由δH 3.24(dd, J=9.2, 4.4 Hz)增大至δH 4.48(dd, J=9.2, 7.2 Hz);同时,化合物1a增加了4个质子信号δH 2.59(4H, m)。13C NMR(100 MHz,CD3OD)δ:174.2, 135.9, 135.7, 133.2, 123.6, 82.6, 74.9, 52.1, 50.7, 46.0, 44.2, 39.0, 38.2, 36.5, 32.3, 32.1, 30.7, 30.1, 28.5, 28.5, 27.6, 26.1, 25.1, 24.7, 22.1, 19.7, 19.3, 18.0, 17.1, 16.2, 13.1。
化合物1b:白色粉末(30 mg),回收率11.1%,HRESIMS确定其分子式为C34H54O5([M+Na]+, m/z 565.386 6; calcd 565.386 9)。IR(KBr)νmax: 3 448, 2 924, 1 726, 1 454, 1377, 1 169, 1 058 cm-1。1H NMR(400 MHz,CD3OD)数据见表 1,经与原料化合物1对照,并参考文献[11],22-H的化学位移由δH 3.68(1H, m)增大至δH 4.90(1H, m);同时,化合物1b增加了4个质子信号δH 2.55(4H, m)。13C NMR(100 MHz,CD3OD)δ:174.0, 136.2, 135.6, 134.4, 121.9, 79.7, 78.6, 52.0, 50.7, 46.1, 41.2, 40.0, 38.3, 37.0, 32.3, 32.0, 30.9, 28.7, 28.5, 28.0, 27.7, 27.7, 26.0, 24.7, 22.1, 19.7, 19.5, 18.0, 16.2, 16.2, 13.8。
化合物1c:白色粉末(103 mg),回收率32.2%,HRESIMS确定其分子式为C38H58O8([M+Na]+, m/z 665.402 8; calcd 665.403 0)。IR(KBr)νmax: 3 385, 2 946, 2 836, 1 731, 1 371, 1 171, 1 024 cm-1。1H NMR(400 MHz,CD3OD)数据见表 1,经与原料化合物1对照,并参考文献[11],3-H的化学位移由δH 3.24(dd, J=9.2, 4.4 Hz)增大至δH 4.48(dd, J=8.8, 7.6 Hz),同时,22-H的化学位移由δH 3.68(1H, m)增大至δH 4.91(1H, m);此外,化合物1c增加了8个质子信号δH 2.57(8H, m)。13C NMR(100 MHz,CD3OD)δ:176.0, 176.0, 174.0, 173.8, 135.8, 135.7, 134.5, 121.9, 82.5, 78.5, 52.0, 50.7, 46.1, 41.2, 39.0, 38.2, 36.5, 32.2, 32.0, 30.6, 29.9, 28.5, 28.0, 27.6, 27.6, 26.1, 25.1, 24.8, 22.1, 19.7, 19.3, 18.1, 17.1, 16.2, 13.8。
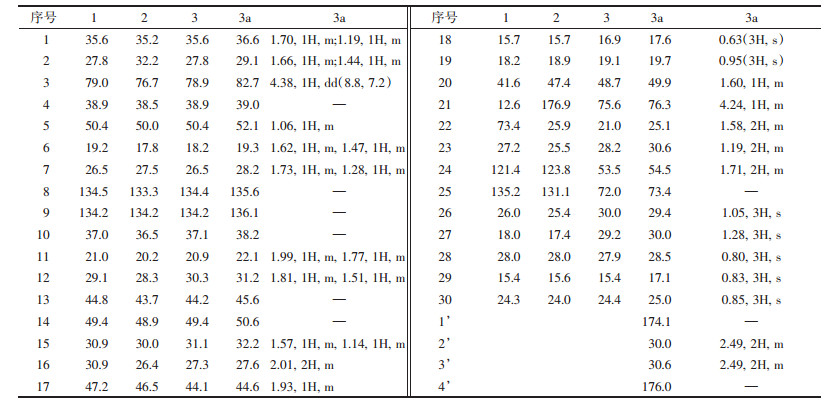
化合物2a:白色粉末(150 mg),回收率38.4%,HRESIMS确定其分子式为C34H52O6([M-H]-, m/z 555.368 8; calcd 555.368 6)。IR(KBr)νmax: 2 950, 1 716, 1 657, 1 375, 1 266, 1 179 cm-1。1H NMR(400 MHz,CD3OD)数据见表 3,经与原料化合物2对照,并参考文献[11],3-H的化学位移由δH 3.00(1H, m)增大至δH 4.39(1H, m);同时,化合物2a增加了4个质子信号δH 2.50(4H, m)。13C NMR(100 MHz,CD3OD)δ:180.5, 176.0, 174.1, 135.7, 135.7, 133.0, 124.9, 82.6, 52.0, 50.7, 49.3, 48.5, 45.5, 39.0, 38.2, 36.5, 33.8, 31.5, 30.6, 30.1, 29.9, 28.5, 28.2, 27.5, 27.0, 26.0, 25.1, 24.8, 22.0, 19.7, 19.2, 17.8, 17.1, 16.5。
化合物3a:白色粉末(4 mg),回收率27.4%,mp 149.5~150.6 ℃,HRESIMS确定其分子式为C34H54O6([M+Na]+, m/z 581.381 7; calcd 581.381 8)。IR(KBr)νmax: 3 436, 2 952, 1 645, 1 452, 1 372, 1 160, 1 017, 667 cm-1。1H NMR(400 MHz,CD3OD)数据见表 3,与原料化合物3对照显示,3-H的化学位移由δH 3.24(dd, J=11.6, 4.4 Hz)增大至δH 4.38(dd, J=8.8, 7.2 Hz);同时,化合物3a增加了4个质子信号δH 2.49(4H, m)。13C NMR(100 MHz,CD3OD)δ:176.0, 174.1, 136.1, 135.6, 82.7, 76.3, 73.4, 54.5, 52.1, 50.6, 49.9, 45.6, 44.6, 39.0, 38.2, 36.6, 32.2, 31.2, 30.6, 30.6, 30.0, 30.0, 29.4, 29.1, 28.5, 28.2, 27.6, 25.1, 25.0, 22.1, 19.7, 19.3, 17.6, 17.1。为证实inonotusane A(3)反应生成3a后,其侧链上的五元环构型未发生变化,通过HSQC谱对目标化合物3a的1H NMR和13C NMR进行归属后(表 2),进一步分析其NOESY谱(图 2)可见H-17/H3-30相关峰,表明H-17为α-构型;另外可见H-21/H-20和H-21/ H3-18相关峰,而未见H-20/H-17相关峰,证实了21β-H和22β-H;同时H-21/H-24相关峰证明H-24为β-构型。化合物1、2、3、1a、1b、1c、2a、3a的结构见图 1。

|
| 图 2 化合物3a的17-侧链的NOESY相关情况 Fig. 2 Key NOESY correlations of 17-side chain in 3a |
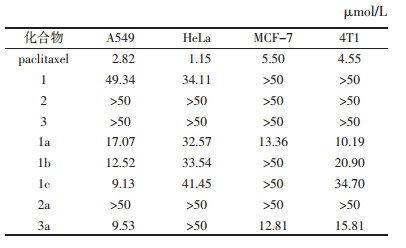
MTT实验结果如表 4所示,化合物1的3-位、22-位酰化后(1a, 1b, 1c)对A549和4T1肿瘤细胞的抑制活性均增强,此外,而其3-位酰化后(1a)还大大增强了对MCF-7肿瘤细胞的抑制活性。化合物2酰化后对活性无影响。化合物3的3-位酰化后对A549、MCF-7及4T1细胞的抑制活性均明显增强,其中对A549表现出最强的抗肿瘤活性(IC50 = 9.53 μmol/L)。
|
所有化合物在结构修饰前后对HeLa细胞的抑制活性均未发生变化,而对其他肿瘤细胞的抑制活性却明显增强(2a除外),推测HeLa细胞对于化合物结构中引入此类酰基并不敏感。2和2a对所有肿瘤细胞均无抑制活性,说明化合物的酰化并不能增强活性,应尝试其他的结构修饰方式。1a、1b、1c和3a均比其原料化合物具更强的抗肿瘤活性,故inotodiol(1)的3位-及22-位、inonotusane A(3)的3-位均有进一步研究的价值,但因他们均为桦褐孔菌的独有成分,目前尚未发现其在其他天然产物中存在。因此,原料问题是其结构修饰工作的关键之一。
天然化合物的结构修饰往往是获得理想活性物质的一个有效手段,其中,三萜类化合物所表现出的广泛的抗肿瘤活性使其成为重要的先导化合物来源,如对鼠纤维肉瘤细胞Meth-A、Lewis型肺癌细胞LLC、乳腺管癌细胞T-47D、恶性肉瘤细胞S180和肝癌细胞Hep G2等都具有较强的细胞毒性,能抑制肝癌细胞的生长并诱导细胞发生凋亡[12-15]。对于三萜类先导化合物的结构修饰及抗肿瘤构效关系研究也受到关注,如同为羊毛脂烷型三萜的灵芝酸的抗肿瘤构效关系研究表明,灵芝酸A、F和H对乳腺癌细胞MDA-MB-231转移的抑制作用,得出结论:C-3、C-7和C-15位置的羟基化有助于提高该类三萜结构对乳腺癌细胞转移的抑制活性[16]。
因此对桦褐孔菌三萜的结构修饰及构效关系研究任重而道远,也希望本研究能为抗肿瘤药物的研究进程贡献新力量。
| [1] | Lemont B. KIER. Triterpenes of Poria obliqua[J]. Eur J Med Chem, 1961, 6 (6): 471–474. |
| [2] | Handa N, Yamada T, Tanaka R. An unusual lanostane-type triterpenoid, spiroinonotsuoxodiol, and other triterpenoids from Inonotus obliquus[J]. Phytochemistry, 2010, 71 (14): 1774–1779. |
| [3] | Lee SH, Hwang HS, Yun J W. Antitumor activity of water extract of a mushroom, Inonotus obliquus, against HT-29 human colon cancer cells[J]. Phytother Res:PTR, 2009, 23 (12): 1784–1789. DOI:10.1002/ptr.v23:12 |
| [4] | Youn MJ, Kim JK, Park SY, et al. Potential anticancer properties of the water extract of Inonotus[corrected] obliquus by induction of apoptosis in melanoma B16-F10 cells[J]. J Ethnopharmacol, 2009, 121 (2): 221–228. DOI:10.1016/j.jep.2008.10.016 |
| [5] | Liu C, Zhao C, Pan HH, et al. Chemical constituents from Inonotus obliquus and their biological activities[J]. J Nat Prod, 2014, 77 (1): 35–41. DOI:10.1021/np400552w |
| [6] | Handa N, Yamada T, Tanaka R. An unusual lanostane-type triterpenoid, spiroinonotsuoxodiol, and other triterpenoids from Inonotus obliquus[J]. Phytochemistry, 2010, 71 (14-15): 1774–1779. DOI:10.1016/j.phytochem.2010.07.005 |
| [7] | Handa N, Yamada T, Tanaka R. Four new lanostane-type triterpenoids from Inonotus obliquus[J]. Phytochem Lett, 2012, 5 (3): 480–485. DOI:10.1016/j.phytol.2012.04.010 |
| [8] | Shin Y, Tamai Y, Terazawa H. Chemical constituents of Inonotus obliquus(Pers:Fr.) Pil.(Aphyllophoromycetideae) Ⅲ:a new triterpene, 3β, 22, 25-trihydroxy-lanosta-8-ene from sclerotia[J]. Int J Med Mushrooms, 2000, 2 (3): 201–207. |
| [9] | Kahlos K, Hiltunen RV, Schantz M. 3β-Hydroxylanosta-8, 24-dien-21-al, a new triterpene from Inonotus obliquus[J]. Planta Med, 1984, 50 (2): 197–198. DOI:10.1055/s-2007-969674 |
| [10] | Zhao FQ, Mai QQ, Ma JH, et al. Triterpenoids from Inonotus obliquus and their antitumor activities[J]. Fitoterapia, 2015, 101 : 34–40. DOI:10.1016/j.fitote.2014.12.005 |
| [11] | Zhao FQ, Xia GY, Chen LX, et al. Chemical constituents from Inonotus obliquus and their antitumor activities[J]. J Nat Med, 2016, 70 (4): 721–730. DOI:10.1007/s11418-016-1002-4 |
| [12] | Min BS, Gao JJ, Nakamura N, et al. Triterpenes from the spores of Ganoderma lucidum and their cytotoxicity against Meth-A and LLC tumor cells[J]. Chem Pharm Bull, 2000, 48 : 1026–1033. DOI:10.1248/cpb.48.1026 |
| [13] | Gao JJ, Min BS, Ahn EM, et al. New trierpene aldehydes, lucialdehydes A-C, from Ganoderma lucidum and their cytotoxicity against murine and human tumor cells[J]. Chem Pharm Bull, 2002, 50 (6): 837–840. DOI:10.1248/cpb.50.837 |
| [14] | Wu TS, Shi LS, Kuo SC. Cytotoxicity of Ganoderma lucidum triterpenes[J]. J Nat Prod, 2001, 64 (8): 1121–1122. DOI:10.1021/np010115w |
| [15] | Li CH, Chen PY, Chang UM, et al. Ganoderic acid X, a lanostanoid triterpene, inhibits topoisomerases and induces apoptosis of cancer cells[J]. Life Sci, 2005, 77 (3): 252–265. DOI:10.1016/j.lfs.2004.09.045 |
| [16] | Jiang J, Grieb B, Thyagarajan A, et al. Ganoderic acids suppress growth and invasive behavior of breast cancer cells by modulating AP-1 and NF-κB signaling[J]. Int J Mol Med, 2008, 21 (5): 577–584. |
2. College of Pharmacy, Henan University, Kaifeng 475004, China