 2020, Vol. 37
2020, Vol. 37文章信息
- 谢慧, 何佶彦
- XIE Hui, HE Jiyan
- 木犀草素纳米混悬剂的制备与质量评价
- Preparation and quality evaluation of luteolin nanosuspension
- 天津中医药, 2020, 37(6): 711-716
- Tianjin Journal of Traditional Chinese Medicine, 2020, 37(6): 711-716
- http://dx.doi.org/10.11656/j.issn.1672-1519.2020.06.25
-
文章历史
- 收稿日期: 2020-03-22

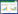



木犀草素(Luteolin)最早是从木犀草科草本植物木犀草(Resedaodorata L.)中分离出来的一种黄酮类化合物,目前已发现存在于菊科、唇形科、天南星科、十字花科、马鞭草科等植物中。最初研究发现木犀草素具有以抗炎,抗氧化,保护神经系统,改善记忆力等药理作用[1],随着研究的深入,发现对木犀草素具有抑制肿瘤细胞增殖,诱导肿瘤细胞凋亡等药理活性[2]。然而,木犀草素在水中溶解度极低(约为61 μg/mL),影响药物体内吸收,口服生物利用度较低[3-4],限制了临床应用。为了提高木犀草素的溶解度及药效,研究人员将其制备成聚合物纳米粒[5]、白蛋白纳米粒[6]、脂质体[7]、固体分散体[8]等新型给药系统。纳米混悬剂是近年来开发的一种纳米给药系统,由于其粒径较小,存在巨大的比表面积和界面能,能够显著增加难溶性药物的溶解度及口服生物利用度[9-10]。因此,本研究将木犀草素制备成纳米混悬剂,通过单因素实验优化了制备工艺参数并确定了处方中稳定剂和表面活性剂的种类,并最终采用Box-Behnken实验设计优化得到木犀草素纳米混悬剂的最优处方,为木犀草素的动物体内药动学研究奠定实验基础。
1 仪器与材料ESW-1.0小型湿法研磨机(上海易勒机电设备有限公司);氧化锆(ZrO2)珠研磨介质(直径为0.5 mm);Zetasizer Nano ZS90纳米粒径/电位分析仪(英国马尔文公司);JSM-7800F扫描电镜(日本电子公司);iChrom 5100系列高效液相色谱仪(大连依利特分析仪器有限公司);RCY-1400T智能溶出试验仪(天津市瑞斯德科技有限公司)。
木犀草素原料药(西安唯奥生物科技有限公司,纯度:98.5%,批号:20180923);木犀草素对照品(成都曼思特生物科技有限公司,纯度:99.5%,批号:MUST-18061407);羟丙基纤维素(HPC SL,日本信越化学);聚维酮(PVP K30,巴斯夫辅料公司);羟丙甲纤维素(HPMC E5,陶氏化学);泊洛沙姆(Poloxamer 188,巴斯夫辅料公司);维生素E聚乙二醇1000琥珀酸酯(TPGs,巴斯夫辅料公司);吐温80(Tween-80,南京威尔药业股份有限公司)。
2 方法与结果 2.1 木犀草素纳米混悬剂的制备通过介质碾磨法[11]制备木犀草素纳米混悬剂。称取一定量的稳定剂(HPC SL、PVP K30或HPMC E5)加入到纯化水中溶解,再加入表面活性剂(Poloxamer 188、Tween-80或TPGs)溶解,备用。称取一定量木犀草素加入到上述溶液中,分散,并加入到介质碾磨机中,加入一定量ZrO2,以一定的速度进行碾磨一定时间,取样测定粒径分布和Zeta电位,制备木犀草素纳米混悬剂。
2.2 粒径分布及Zeta电位测定取木犀草素纳米混悬剂加入少量蒸馏水稀释,用Zetasizer Nano ZS90纳米粒径/电位分析仪测定木犀草素纳米混悬剂的粒径分布和Zeta电位,每份样品重复测定3次。
2.3 处方工艺筛选 2.3.1 稳定剂种类筛选文献报道[12]纳米混悬剂处方中加入稳定剂及表面活性剂可以提高体系的稳定性。本研究固定药物浓度为30 mg/mL,碾磨介质与混悬剂体积之比为1:1,碾磨速度为2 500 r/min,碾磨时间为4 h,每个处方中分别加入浓度为2%的HPC SL、PVP K30和HPMC E5作为稳定剂制备木犀草素纳米混悬剂,测定粒径分布及Zeta电位,筛选稳定剂。研究结果表明,不同种类的稳定剂会对木犀草素纳米混悬剂的粒径及Zeta电位大小产生一定的影响,使用HPC SL作为稳定剂制备的纳米混悬剂粒径分布最小,Zeta电位较低,因此本研究使用HPC SL作为稳定剂制备木犀草素纳米混悬剂。
2.3.2 稳定剂浓度筛选固定药物浓度为30 mg/mL,碾磨介质与混悬剂体积之比为1:1,碾磨速度为2 500 r/min,碾磨时间为4 h,在处方中分别加入浓度为0.5、0.75、1、2、5%的HPC SL制备木犀草素纳米混悬剂,测定粒径分布及Zeta电位,筛选稳定剂浓度。研究验结果表明,稳定剂浓度对木犀草素纳米混悬剂的粒径及Zeta电位产生一定的影响,随着稳定剂浓度的增大,制备的纳米混悬剂粒径增大,Zeta电位随着稳定剂的浓度增加而增大,因此稳定剂的浓度需要进一步优化。
2.3.3 表面活性剂种类筛选文献报道[13]纳米混悬剂中的表面活性剂种类会对其粒径及稳定性产生一定的影响。固定药物浓度为30 mg/mL,碾磨介质与混悬剂体积之比为1:1,碾磨速度为2 500 r/min,碾磨时间为4 h,HPC SL浓度为2%,处方中分别加入浓度为0.2%的Poloxamer 188、TPGs和Tween-80作为表面活性剂制备木犀草素纳米混悬剂,测定粒径分布及Zeta电位,筛选表面活性剂。研究结果表明,不同种类的表面活性剂会对木犀草素纳米混悬剂的粒径及Zeta电位大小产生一定的影响,使用TPGs作为表面活性剂制备的纳米混悬剂粒径分布最小,Zeta电位较低,因此本研究使用TPGs作为表面活性剂制备木犀草素纳米混悬剂。
2.3.4 表面活性剂浓度筛选固定药物浓度为30 mg/mL,碾磨介质与混悬剂体积之比为1:1,碾磨速度为2 500 r/min,碾磨时间为4 h,HPC SL浓度为2%,在处方中分别加入浓度为0.1、0.15、0.2、0.25、0.3%的TPGs制备木犀草素纳米混悬剂,测定粒径分布及Zeta电位,筛选表面活性剂浓度。研究结果表明,表面活性剂浓度对木犀草素纳米混悬剂的粒径影响不大,但对Zeta电位影响较为显著,随着TPGs浓度的增大,Zeta电位升高,因此表面活性剂浓度需要进一步优化。
2.3.5 药物浓度筛选固定碾磨介质与混悬剂体积之比为1:1,碾磨速度为2 500 r/min,碾磨时间为4 h,HPC SL浓度为2%,TPGs浓度为0.2%,药物浓度分别为10、20、30、40 mg/mL制备木犀草素纳米混悬剂,测定粒径分布及Zeta电位,筛选表面活性剂。研究结果表明,药物浓度对木犀草素纳米混悬剂的粒径具有一定影响,随着药物浓度的增大,纳米混悬剂的粒径先减小后增大,而药物浓度对Zeta电位影响不显著,因此药物浓度需要进一步优化。
2.3.6 碾磨介质用量筛选固定药物浓度为30 mg/mL,碾磨速度为2 500 r/min,碾磨时间为4 h,HPC SL浓度为2%,TPGs浓度为0.2%,分别选择碾磨介质与混悬剂体积之比为0.2:1,0.4:1,0.6:1,0.8:1,1:1,制备木犀草素纳米混悬剂,测定粒径分布及Zeta电位,筛选碾磨介质用量。研究结果表明,碾磨介质体积对木犀草素纳米混悬剂的粒径具有一定的影响,随着碾磨介质体积的增加,制备的纳米混悬剂粒径逐渐减小,碾磨介质体积对Zeta电位影响不显著,因此本研究确定碾磨介质体积与混悬剂体积之比为1:1。
2.3.7 碾磨时间筛选固定药物浓度为30 mg/mL,碾磨介质与混悬剂体积之比为1:1,碾磨速度为2 500 r/min,HPC SL浓度为2%,TPGs浓度为0.2%,分别选择碾磨时间为1、2、4、6 h,制备木犀草素纳米混悬剂,测定粒径分布及Zeta电位,筛选碾磨时间。研究结果表明,碾磨时间对木犀草素纳米混悬剂的粒径产生一定的影响,对Zeta电位影响不显著,随着碾磨时间的增加,制备的纳米混悬剂粒径逐渐减小,但是碾磨时间达到6 h,粒径不再减小,因此确定碾磨时间为4 h。
2.3.8 碾磨速度筛选固定药物浓度为30 mg/mL,碾磨介质与混悬剂体积之比为1:1,碾磨时间为4 h,HPC SL浓度为2%,TPGs浓度为0.2%,分别选择碾磨速度为1 000、1 500、2 000、2 500、3 000 r/min,制备木犀草素纳米混悬剂,测定粒径分布及Zeta电位,筛选碾磨速度。研究结果表明,碾磨速度对木犀草素纳米混悬剂的粒径产生一定的影响,对Zeta电位影响不显著,随着碾磨速度的增加,制备的纳米混悬剂粒径逐渐减小,但是碾磨速度超过2 500 r/min,粒径不再减小,因此确定碾磨速度为2 500 r/min。
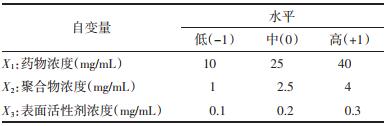
2.4 实验设计 2.4.1 Box-Behnken实验设计优化根据单因素实验筛选结果,以药物浓度(X1),稳定剂浓度(X2)和表面活性剂浓度(X3)作为考察因素,以木犀草素纳米混悬剂的粒径分布(Y1)布和Zeta电位(Y2)作为评价指标,通过Box-Behnken实验设计优化木犀草素纳米混悬剂的处方,3个变量水平见表 1,生成的15组实验方案及结果见表 2。
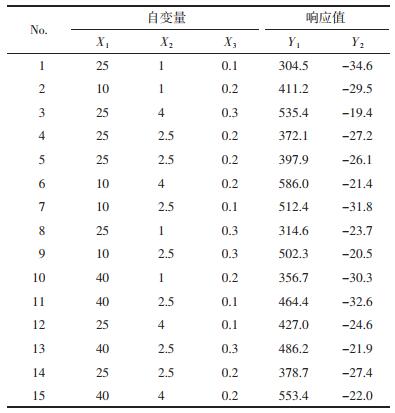
通过“Box-Behnken实验设计”统计分析实验结果见表 3,两个模型P值均为0.000 8,小于0.05,表明两个拟合模型均显著;模型中R2和矫正R2用于估模型方程的可接受性,Y1的R2值和矫正R2值分别为0.982 2和0.950 2,Y2的R2值和矫正R2值分别为0.981 4和0.947 8,之间差值均小于0.1,说明两个模型可信度较高;失拟项P值分别为0.254 6和0.236 1,均大于0.05,说明两个模型的预测值与实际值之间无统计学差异,模型预测性较高。Y1和Y2响应值与3个自变量之间的相关性均符合多元二次方程模型。
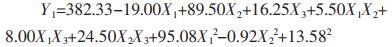 |
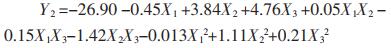 |
另外,通过方差分析结果可知3个变量与响应值之间的关系,即:X1、X2、X2X3、X12对粒径分布(Y1)具有显著影响(P < 0.05),而X2、X3、X2X3对Zeta电位(Y2)具有显著影响(P < 0.05)。自变量与响应值之间的关系见图 1、2。

|
| 图 1 药物浓度(X1),稳定剂浓度(X2)、表面活性剂浓度(X3)对木犀草素纳米混悬剂的粒径分布(Y1)的效应面图 Fig. 1 Effect of luteolin concentration (X1), stabilizer concentration (X2), surfactant concentration (X3) on the particle size distribution (Y1) of luteolin nanosuspension |

|
| 图 2 药物浓度(X1),稳定剂浓度(X2)、表面活性剂浓度(X3)对木犀草素纳米混悬剂的Zeta电位(Y2)的效应面图 Fig. 2 Effect of luteolin concentration (X1), stabilizer concentration (X2), surfactant concentration (X3) on Zeta potential (Y2) of luteolin nanosuspension |
由效应面图 1可知,木犀草素纳米混悬剂的粒径随着药物浓度的增加先减小后增大,随着稳定剂浓度的增加粒径增大,表面活性剂浓度对粒径的影响可以忽略不计。
由效应面图 2可知,木犀草素纳米混悬剂的Zeta电位随着稳定剂浓度的增加而升高,也随着表面活性剂浓度的增加而升高,药物浓度对Zeta电位的影响可以忽略不计。
2.4.3 模型预测与验证本研究确定木犀草素纳米混悬剂的处方应具有粒径分布“最小”,Zeta电位“最小”,通过“Box-Behnken实验设计”优化得到木犀草素纳米混悬剂的最优处方组成为:药物浓度为28.0 mg/mL,稳定剂浓度为1.5 mg/mL,表面活性剂浓度为0.2 mg/mL,软件预测木犀草素纳米混悬剂的粒径为332.2 nm,Zeta电位为-30.5 mV。根据优化的木犀草素纳米混悬剂处方制备3批样品,经测定粒径为(324.3±21.6)nm,Zeta电位为(-31.4±0.9)mV,实测值与预测值接近,模型预测可靠性较高。
2.5 微观形态取木犀草素纳米混悬剂加入少量蒸馏水稀释,取1滴样品滴加到载玻片上,挥干水分,喷金,放在扫描电镜下观察木犀草素纳米混悬剂的微观形态。扫描电镜照片显示木犀草素纳米混悬剂呈颗粒状均匀分布,粒径大约在100~500 nm之间。见图 3。

|
| 图 3 木犀草素纳米混悬剂扫描电镜(×20 000) Fig. 3 The scanning electron microscope of luteolin nanosuspension (×20 000) |
使用桨法测定木犀草素纳米混悬剂与木犀草素原料药的体外溶出度。溶出介质为0.5%(w/w)吐温-80磷酸盐缓冲溶液(pH6.8),体积为500 mL,桨板转速为50 r/min,水浴温度为37 ℃。开启溶出仪,用移液器精密移取0.5 mL木犀草素纳米混悬剂加入到溶出杯中,分别在5、10、20、30、45、60 min取样5 mL溶出介质,20 000 r/min高速离心,取1 mL上清液经适当稀释,使用HPLC法[14]测定药物含量;另取15 mg木犀草素原料药加入到溶出杯中,按照上述方法操作,测定药物含量。通过绘制时间-溶出度曲线比较木犀草素纳米混悬剂与木犀草素原料药的溶出度。见图 4。

|
| 图 4 木犀草素纳米混悬剂与木犀草素原料药体外溶出曲线(n=6) Fig. 4 In vitro release curve of luteolin nanosuspension and luteolin (n=6) |
由木犀草素纳米混悬剂与木犀草素原料药溶出曲线可知,木犀草素原料药在10 min累积溶出度为18.3%,30 min累积溶出度为34.4%,60 min累积溶出度为42.2%,体外累积溶出度数据经威布尔方程拟合为:lnln[1/(1-C(t))]=0.6054ln(t)-2.994 9(R2=0.988 9),经计算体外累积溶出度50%的时间为77.6 min,累积溶出度63.2%的时间为89.6 min;而木犀草素纳米混悬剂在10 min溶出度为76.4%,30 min溶出度为96.3%,60 min溶出度98.7%,体外累积溶出度数据经威布尔方程拟合为:lnln[1/(1-C(t))]=0.771 8ln(t)-1.573 3(R2=0.971),经计算体外累积溶出度50%的时间为4.8 min,累积溶出度63.2%的时间为7.7 min,表明木犀草素纳米混悬剂能够显著提高药物溶出度。
3 讨论目前,药物纳米粒子的制备技术从原理上可分为“自上而下”(Top-down)和“自下而上”(Bottom-up)两种制备技术[15]。“自下而上”分为反溶剂沉淀法和化学反应沉淀法,“自上而下”分为介质碾磨法、高压均质法和微射流法,由于介质碾磨法在纳米混悬剂制剂生产中具有高度的可重现性、可避免碾磨腔体递质污染、易于规模放大,因此本研究选用介质碾磨法制备木犀草素纳米混悬剂。
纳米混悬剂属于热力学不稳定系统,在处方中需要添加稳定剂以降低液-固界面张力,提高体系的稳定性。本研究在木犀草素纳米混悬剂的处方中加入HPC SL和TPGs作为稳定剂,HPC SL可以吸附到纳米混悬剂表面,形成一层薄膜,阻碍颗粒之间的聚集,起到空间位阻作用,而表面活性剂TPGs可降低了药物与水之间的界面张力,润湿药物表面,通过稳定剂与表面活性物质的双重作用提高了木犀草素纳米混悬剂的稳定性。
| [1] |
王继双, 何焱, 张文静, 等. 木犀草素的药理作用研究进展[J]. 生命科学, 2013, 25(6): 560-565. WANG J S, HE Y, ZHANG W J, et al. Advances in studies on pharmacological effects of luteolin[J]. Chinese Bulletin of Life Sciences, 2013, 25(6): 560-565. |
| [2] |
张芳芳, 沈汉明, 朱心强. 木犀草素抗肿瘤作用的研究进展[J]. 浙江大学学报(医学版), 2006, 35(5): 573-578. ZHANG F F, SHEN H M, ZHU X Q. Research progress on anti-tumor effects of luteolin[J]. Journal of Zhejiang University (Medical Sciences), 2006, 35(5): 573-578. DOI:10.3785/j.issn.1008-9292.2006.05.021 |
| [3] |
KAMINAGA Y, NAGATSU A, AKIYAMA T. Production of unnatural glucosides of curcumin with drastically enhanced water solubility by cellsuspension cultures of Catharanthus roseus[J]. FEBS Letters, 2003, 555(2): 311-316. DOI:10.1016/S0014-5793(03)01265-1 |
| [4] |
ZHOU P, LI L P, LUO S Q. Intestinal absorption of luteolin from peanut hull extract is more efficient than that from individual pure luteolin[J]. Journal of Agricultural and Food Chemistry, 2008, 56(1): 296-300. DOI:10.1021/jf072612+ |
| [5] |
王萌, 张淼, 张斐娜, 等. 木犀草素-mPEG-PLGA载药纳米粒的构建、优化与体外抗肿瘤活性研究[J]. 武警后勤学院学报(医学版), 2018, 27(11): 900-905, 910. WANG M, ZHANG M, ZHANG F N, et al. Construction, optimization and antitumor activity of luteolin-loaded m PEG-PLGA nanoparticles[J]. Journal of Logistics University of PAP (Medical Sciences), 2018, 27(11): 900-905, 910. |
| [6] |
付伟, 柴栋, 文颖, 等. 甘草酸偶联牛血清白蛋白载木犀草素纳米粒制备及其体外活性研究[J]. 热带医学杂志, 2018, 18(1): 22-27. FU W, CHAI D, WEN Y, et al. Preparation and in vitro activity of glycyrrhizic acid coupled bovine serum albumin loaded with luteolin nanoparticles[J]. Journal of Tropical Medicine, 2018, 18(1): 22-27. DOI:10.3969/j.issn.1672-3619.2018.01.006 |
| [7] |
WU G, LI J, YUE J, et al. Liposome encapsulated luteolin showed enhanced antitumor efficacy to colorectal carcinoma[J]. Molecular Medicine Reports, 2018, 17(2): 2456-2464. |
| [8] |
KHAN J, ALEXANDER A, AJAZUDDIN, et al. Luteolin-phospholipid complex:preparation, characterization and biological evaluation[J]. Journal of Pharmacy and Pharmacology, 2014, 66(10): 1451-1462. DOI:10.1111/jphp.12280 |
| [9] |
GHOSH I, BOSE S, VIPPAGUNTA R, et al. Nanosuspension for improving the bioavailability of a poorly soluble drug and screening of stabilizing agents to inhibit crystal growth[J]. International Journal of Pharmaceutics, 2011, 409(1-2): 260-268. DOI:10.1016/j.ijpharm.2011.02.051 |
| [10] |
AHUJA B K, JENA S K, PAIDI S K. Formulation, optimization and in vitro-in vivo evaluation of febuxostat nanosuspension[J]. International Journal of Pharmaceutics, 2015, 478(2): 540-552. DOI:10.1016/j.ijpharm.2014.12.003 |
| [11] |
JUHNKE M, MARTIN D, JOHN E. Generation of wear during the production of drug nanosuspensions by wet media milling[J]. European Journal of Pharmaceutics and Biopharmaceutics, 2012, 81(1): 214-222. DOI:10.1016/j.ejpb.2012.01.005 |
| [12] |
HU J, NG W K, DONG Y, et al. Continuous and scalable process for water-redispersible nanoformulation of poorly aqueous soluble APIs by antisolvent precipitation and spray-drying[J]. International Journal of Pharmaceutics, 2011, 404(1-2): 198-204. DOI:10.1016/j.ijpharm.2010.10.055 |
| [13] |
GHOSH I, SCHENCK D, BOSE S, et al. Optimization of formulation and process parameters for the production of nanosuspension by wet media milling technique:Effect of Vitamin E TPGS and nanocrystal particle size on oral absorption[J]. European Journal of Pharmaceutical Sciences, 2012, 47(4): 718-728. DOI:10.1016/j.ejps.2012.08.011 |
| [14] |
陈以军, 任亚硕, 吴德玲. 反相高效液相色谱法测定醉鱼草果实中木犀草素的含量[J]. 安徽中医药大学学报, 2014, 33(4): 87-89. CHEN Y J, REN Y S, WU D L. Determination of luteolin in fruit of buddleja lindleyana Fort. by reverse phase-high performance liquid chromatography[J]. Journal of Anhui Traditional Chinese Medical College, 2014, 33(4): 87-89. DOI:10.3969/j.issn.2095-7246.2014.04.028 |
| [15] |
MOSCHWITZER J P. Drug nanocrystals in the commercial pharmaceutical development process[J]. International Journal of Pharmaceutics, 2013, 453(1): 142-156. DOI:10.1016/j.ijpharm.2012.09.034 |