 2025, Vol. 42
2025, Vol. 42文章信息
- 张婷婷, 张悦, 邢志美, 等.
- ZHANG Tingting, ZHANG Yue, XING Zhimei, et al.
- 大花水蓑衣叶绿体基因组特征及系统发育分析
- Characteristics of chloroplast genome and phylogenetic analysis of Hygrophila megaliths Merr.
- 天津中医药, 2025, 42(5): 638-646
- Tianjin Journal of Traditional Chinese Medicine, 2025, 42(5): 638-646
- http://dx.doi.org/10.11656/j.issn.1672-1519.2025.05.15
-
文章历史
- 收稿日期: 2024-12-20





大花水蓑衣(Hygrophila megalantha Merr.)是爵床科水蓑衣属的植物,其种子可作药用,称“广天仙子”,收载于《儿茶等43种进口药材质量标准》[1]。具有清热泻火,凉血解毒的功效,可外用治热毒、疮疖。广天仙子主要产自越南、缅甸等国家,属于进口药材。现代药理研究表明其可以治疗急性乳腺炎、褥疮、压疮、化脓性感染创面以及药物外渗至组织损伤或静脉炎[2-6]。
尽管目前对广天仙子药理作用的研究逐渐深入,针对其原植物大花水蓑衣的基原研究较少。而叶绿体基因组因其基因序列较为保守和在植物进化中的重要作用而被广泛用于系统进化分析和物种鉴定[7-8]。随着高通量测序技术的快速发展,可用叶绿体基因组的数量不断增多,为药用植物叶绿体基因组研究提供了重要依据。故该研究对大花水蓑衣及其同属近源物种Hygrophila sp.的叶绿体基因组进行组装和分析,旨在分析系统发育位置和进化关系提供研究依据。
1 材料该实验的3批样品均由天津市药检所提供,由中国科学院华南植物园的邓云飞教授进行鉴定,确定种子样品GTXZ-2、GTXZ-3为广天仙子正品(Hygrophila megalantha),GTXZ-1为同属近源物种(Hygrophila sp.)。
2 方法 2.1 总DNA提取和测序采用天根植物基因组DNA提取试剂盒进行DNA提取,并设置空管作为阴性对照以判断实验过程中是否存在污染。使用NanoDropTM分光光度计(Thermo Scientific,美国)对提取得到的DNA进行浓度及纯度检测。用2%的琼脂糖凝胶电泳检测总DNA的质量。随后将样品DNA送至深圳华大基因科技服务有限公司,基于DNBseq测序平台,构建插入片段为350 bp的文库进行PE150测序。
2.2 叶绿体基因组组装对公司返回的测序数据进行质量控制,得到clean data。使用GetOrganelle软件[9]进行叶绿体基因组组装,k-mer设置为21,45,65,85,105。然后在Geneious软件[10](许可证:PRIME-ZCXAG-M6DW2-E5YRY)中将3批样品的clean data分别比对到其对应的叶绿体基因组上,检查reads在叶绿体基因组中的覆盖情况,避免出现组装错误。
2.3 叶绿体基因组注释选择PGA软件[11]、网页注释工具CPG[12]和GeSeq[13]对组装后的3个叶绿体基因组环进行基因功能注释。将3种注释结果导入Geneious中进行比较,以PGA的注释结果作为主要参照,同时结合CPG和GeSeq的注释结果,仔细检查校对每个基因的注释结果。利用“Translation”和“Find ORF”功能检查并手动调整蛋白编码基因的起始和终止密码子。使用tRNAscan-SE软件[14]注释tRNA基因,使用RNAmmer 1.2 Server[15]注释rRNA基因。IR区的注释通过代码:formatdb -i 1.fasta -o F -p F blastn -query 1.fasta -db 1.fasta -out 1 -evalue 10 -outfmt 7实现。
2.4 叶绿体基因组的上传及可视化将注释好的叶绿体基因组序列用Sequin软件[16]提交至NCBI。使用Chloroplot网站[17](https://irscope.shinyapps.io/Chloroplot/)在线绘制叶绿体基因组图谱。
2.5 叶绿体基因组序列分析利用mVISTA[18][mVISTA Input(lbl.gov)]进行叶绿体基因组序列差异分析,对比方式为LAGAN。
2.6 密码子使用偏好性分析使用Geneious提取叶绿体基因组的共有蛋白编码基因并串联。然后将结果文件导入MEGA X软件[19],计算同义密码子相对使用率(RSCU)。
2.7 重复序列分析使用在线网站MISA-web[20][Misa-web-IPK Gatersleben(ipk-gatersleben.de)]进行简单重复序列分析(SSRs)。参数设置:单核苷酸重复次数≥10,二核苷酸重复次数≥5,三核苷酸重复次数≥4、四核苷酸、五核苷酸和六核苷酸重复单元≥3。另,在REPuter[21]在线软件中进行长重复序列分析,包括正向重复序列(Forward repeat)、反向重复序列(Reverse repeat)、互补重复序列(Complementary repeat)和回文重复序列(Palindromic repeat);最小重复单元设置为30,汉明距离(Hamming Distance)设置为3。
2.8 IR/SC区边界收缩和扩张分析使用在线网站IRscope[22](irscope.shinyapps.io/irapp/)绘制叶绿体基因组的IR/SC边界基因分布对比图,分析边界的收缩与扩张特征。
2.9 系统发育分析为探究Hygrophila megalantha的系统发育位置,将Hygrophila megalantha、Hygrophila sp.的叶绿体基因组与NCBI上Hygrophila megalantha所在芦莉草族已发表的叶绿体基因组(见表 1),作为内群用于系统发育树的构建。另外,爵床亚科下芦莉草族外的穿心莲Andrographis paniculata(NC_ 022451)、红花假杜鹃Barleria repens(NC_070083)、刺苞老鼠簕Acanthus leucostachyu(NC_070388)3个物种被设置成外类群。使用PhyloSuite v1.2.1软件[23]对以上叶绿体基因组进行蛋白编码基因的提取、串联和对齐,得到一个超级矩阵作为构建系统发育树的输入数据。使用PhyloSuite v1.2.1软件中的IQ-TREE插件[24]构建Maximum likelihood(ML)系统发育树。通过插件的模型自动选择功能计算最优的核苷酸替代模型,结果为GTR+F+I+G4,重复迭代次数为1 000。

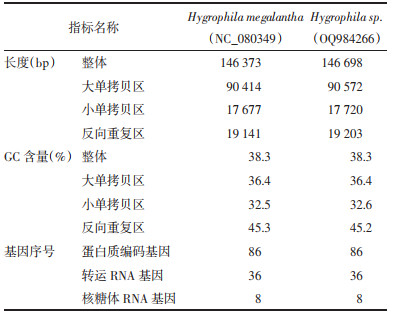
实验组装得到的Hygrophila megalantha和Hygrophila sp.的叶绿体基因组是典型的四分体双链环形结构,包括1个大单拷贝区(LSC),1个小单拷贝区(SSC)和两个等长的反向重复区(IR)。由于3批样本来源于两个基原物种,且正品样本GTXZ-2和GTXZ-3测序结果完全一致,所以接下来的相关分析只使用两个样本(GTXZ-2和GTXZ-1)的叶绿体基因组环完成。两种水蓑衣属植物的叶绿体基因组物理图谱见图 1。Hygrophila megalantha和Hygrophila sp.的叶绿体基因组序列长度分别为146 373 bp和146 698 bp。总体鸟嘌呤和胞嘧啶含量(GC)含量均为38.3%,其中IR区的GC含量最高,其次是LSC区,GC含量最低的是SSC区。两个叶绿体基因组基本特征信息见表 2。

|
| 注:图A,Hygrophila megalantha;图B,Hygrophila sp.。内侧基因顺时针转录,外侧基因逆时针转录,内环中的灰色部分表示GC含量百分比。 图 1 Hygrophila megalantha和Hygrophila sp.的叶绿体基因组物理图谱 Fig. 1 Gene map of chloroplast genomes of Hygrophila megalantha and Hygrophila sp. |
|
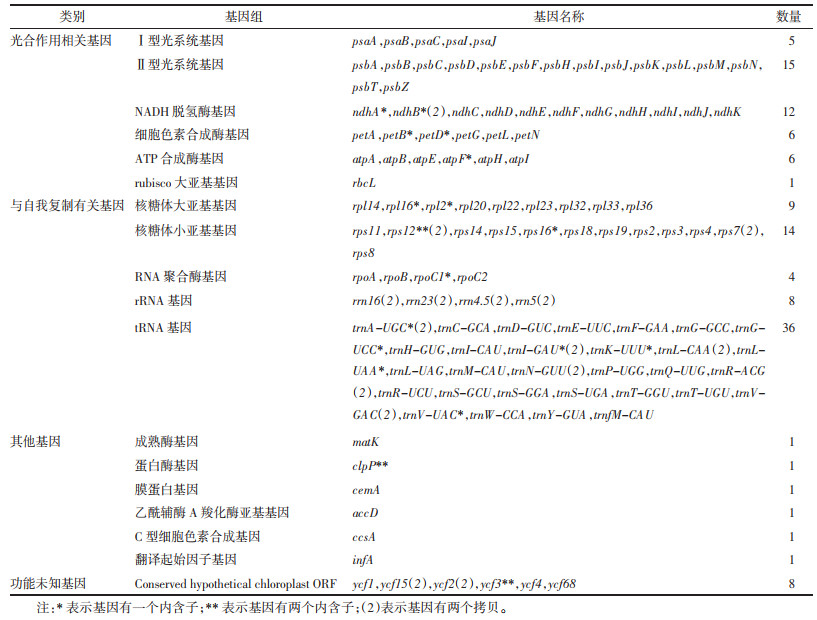
Hygrophila megalantha和Hygrophila sp.的叶绿体基因组编码130个基因,包含蛋白编码基因(CDS)86个、转运RNA(tRNA)基因为36个,核糖体RNA(rRNA)8个。其中有18个基因含有内含子,含有1个内含子的为蛋白编码基因ndhA,ndhB,petB,petD,atpF,rpl16,rpl2,rps16,rpoC1,tRNA基因trnA-UGC,trnG-UCC,trnI-GAU,trnK-UUU,trnL-UAA,trnV-UAC;基因rps12,clpP,ycf3含有两个内含子。
这些基因根据功能主要分为3类:第1类是与光合作用相关的基因,包括5种Ⅰ型光系统基因、15种Ⅱ型光系统基因、11种NADH脱氢酶基因、6种细胞色素合成酶基因、6种ATP合成酶基因以及1种rubisco大亚基基因;第2类基因与自我复制有关,包括9种核糖体大亚基基因、12种核糖体小亚基基因、4种RNA聚合酶基因、4个rRNA基因和30个tRNA基因;第3类是其他功能基因,包括1种成熟酶基因、1种蛋白酶基因、1种膜蛋白基因、1种乙酰辅酶A羧化酶亚基基因、1种C型细胞色素合成基因、1种翻译起始因子基因;另外还有6种功能未知的基因。见表 3。
|
为研究两个叶绿体基因组序列差异程度,该研究以Hygrophila megalantha叶绿体序列为参考,运用mVISTA对Hygrophila megalantha和Hygrophila sp.进行全基因组比较。结果表明(图 2),Hygrophila megalantha和Hygrophila sp.叶绿体基因组的基因结构和排列顺序高度保守。但两者的碱基序列存在一定差异,主要体现在非编码区上。此外,一些蛋白编码基因显示出较高变异,如matK,rpl22,ccsA等,这些高变区可以作为潜在区域用于分子条形码的开发研究。就四分体结构而言,两者在IR区上的变异程度远小于单拷贝区,说明IR区在进化过程中更为保守。

|
| 图 2 Hygrophila megalantha和Hygrophila sp.叶绿体基因组序列比较 Fig. 2 Comparison of chloroplast genome sequences between Hygrophila megalantha and Hygrophila sp. |
某密码子使用次数与其无偏性使用次数的比值即为同义密码子相对使用率(RSCU),无偏性使用次数为该密码子编码的氨基酸所有密码子的平均使用次数,该值排除了氨基酸组成对密码子使用产生的影响,能直观地反映密码子偏性[25]。若RSCU值为1,说明该密码子没有使用偏性;若RSCU值大于1,说明该密码子使用频率相对更高,反之,表明其使用频率较低。
该实验分析了Hygrophila megalantha和Hygrophila sp.叶绿体基因组的密码子使用偏性。结果显示,Hygrophila megalantha和Hygrophila sp.分别有24 580和24 645个密码子。两者的密码子使用偏性大体相似:RSCU值大于1的密码子有30个,除UUG以外,均以碱基A/U结尾;RSCU值小于1的有32个,除CUA、AUA、UGA外,均以碱基C/G结尾;只有密码子AUG(M)和UGG(W)没有使用偏性(图 3)。说明Hygrophila megalantha和Hygrophila sp.叶绿体基因组密码子都倾向以碱基A/U结尾,而以G/C结尾的密码子使用频率较低。

|
| 注:图A,Hygrophila megalantha;图B,Hygrophila sp.。 图 3 两种水蓑衣属植物叶绿体基因组的密码子相对使用度 Fig. 3 Relative synonymous codon usage of two chloroplast genomes in Hygrophila species |
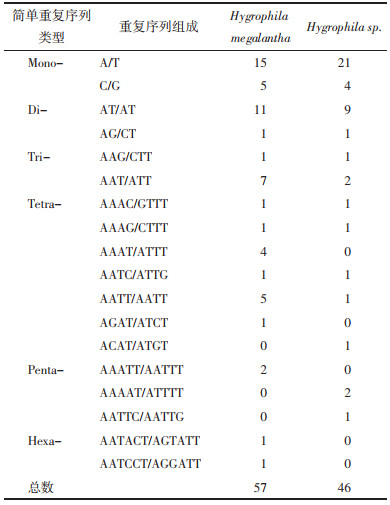
微卫星(Microsatellite),又称为简单重复序列(SSRs)或短串联重复序列(STR),是1种由1~6个重复核苷酸单元组成的串联重复序列[26]。实验使用MISA对Hygrophila megalantha和Hygrophila sp.进行SSRs分析,结果如表 4所示。在Hygrophila megalantha和Hygrophila sp.中分别检测出57和46个微卫星序列。其中单核苷酸重复单元数量最多。观察其碱基类型可以发现,A/T碱基重复最多。
|
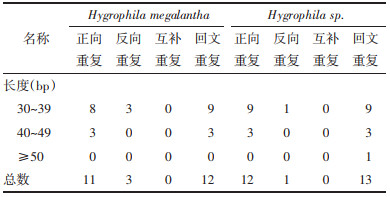
此外,实验还使用REPuter分析了两个物种叶绿体基因组的长重复序列,结果如表 5所示。两个叶绿体基因组均检测到26个长重复序列,包括正向重复、反向重复和回文重复3种类型,没有检测到互补重复。其中正向重复和回文重复比较常见。长重复序列的长度主要集中在30~39 bp。
|
植物叶绿体基因组IR/SC边界的收缩和扩展决定了植物叶绿体基因组的大小[27],是全基因组大小变化的主要机制[28]。IR区与LSC和SSC区存在4个边界,即LSC/IRb、IRb/SSC、SSC/IRa、IRa/LSC,基因进化伴随着4个边界的扩张与收缩。实验对Hygrophila megalantha和Hygrophila sp.的IR/SC边界区域进行比较分析,结果如图 4所示。两个叶绿体基因组边界基因的位置和类型差异较小。从4个区域整体的分布大小来看,两者的主要差异集中于JLA边界:Hygrophila megalantha的trnH基因横跨JLA边界,而Hygrophila sp.的trnH基因位于LSC区内。

|
| 注:JLB、JSB、JSA和JLA分别表示LSC/IRb、IRb/SSC、SSC/IRa和IRa/LSC 4个边界的连接点。 图 4 Hygrophila megalantha和Hygrophila sp.叶绿体基因组IR区的收缩和扩张 Fig. 4 Contraction and expansion of the IR regions in the chloroplast genomes of Hygrophila megalantha and Hygrophila sp. |
为探究Hygrophila megalantha和Hygrophila sp.在爵床科中的系统发育位置,研究采用最大似然法(ML)进行系统发育树的重建。选择Hygrophila megalantha、Hygrophila sp.以及15个芦莉草族下的叶绿体基因组作为内群,选用爵床亚科下芦莉草族外的3个叶绿体基因组作为外类群。结果如图 5所示。在重建的系统发育树中,可以看出:3种爵床亚科芦莉草族外的外类群植物,与内群物种归属不同的分支,可以明显区分开。芦莉草族物种的这一分支上,Echinacanthus属的3个物种黄花恋岩花Echinacanthus lofouensis、龙州恋岩花Echinacanthus longzhouensis和长柄恋岩花Echinacanthus longipes聚为一支;另一分支,由芦莉草属的3个物种蓝花草Ruellia simplex(Ruellia brittoniana)、Ruellia speciosa、艳芦莉Ruellia elegans聚成的小分支,与Echinacanthus attenuatus、Hygrophila megalantha和Hygrophila sp.以及马蓝属的7个物种聚成的小分支共同组成。其中Hygrophila megalantha和Hygrophila sp.聚为一支,亲缘关系最近,支持度为100。两者同马蓝属的7个物种邻近,亲缘关系较近,聚为一个分支,支持度为80。可以发现,在芦莉草族中与Hygrophila megalantha亲缘关系由远及近依次是Echinacanthus属、芦莉草属、马蓝属、水蓑衣属(不考虑Echinacanthus属下的Echinacanthus attenuatus)。

|
| 图 5 围绕Hygrophila megalantha和Hygrophila sp.重建的系统发育树 Fig. 5 Phylogenetic tree reconstructed around Hygrophila megalantha and Hygrophila sp. |
该研究首次报道了Hygrophila megalantha和Hygrophila sp.的叶绿体基因组,与大多数高等植物叶绿体基因组一样,为典型的闭合环状双链结构。从4个部分的序列长度看,Hygrophila megalantha和Hygrophila sp.的LSC区、SSC区及IR区分别相差158 bp、43 bp和62 bp,序列长度差异较小。两个物种叶绿体基因组的总GC含量及各部分GC含量存在细微差异。两者叶绿体基因组在基因数量和类型上一致。此外,两者碱基序列差异主要体现在单拷贝区,而IR区高度保守。
基因组重复序列是研究物种进化进程以及遗传特征的重要依据[29]。本研究主要考察了SSRs和长重复序列。结果显示Hygrophila megalantha和Hygrophila sp.叶绿体基因组SSRs的类型和数目有所差异。其中A/T单核苷酸占主要优势,与叶绿体基因组SSRs的存在方式以A或T单碱基的重复为主[30]的观点一致。SSRs具有物种特异性,因此后续研究可基于检测到的SSRs[31-32]用于Hygrophila megalantha和Hygrophila sp.以及其他混伪品的鉴别。长重复序列是基因组大小变异的主要来源[33],重复序列不固定的比例和位置推动了植物基因组的进化[34]。Hygrophila megalantha和Hygrophila sp.叶绿体基因组的长重复序列类型和数目均相似,主要以正向重复为主,未检测到互补重复;长度多在30~39 bp。
密码子由mRNA上相邻的3个碱基构成,是连接核酸与蛋白质的纽带,在生物遗传信息传递时具有重要作用。研究表明,密码子的使用存在不均等的现象,这一现象称为密码子偏好性(codon usage bias)[35]。RSCU计算结果表明,两种植物的密码子使用偏性相似,倾向以碱基A/U结尾,而以G/C结尾的密码子使用频率较低;只有密码子AUG(M)和UGG(W)没有使用偏性。影响密码子偏好性的因素可能是为自然选择和基因突变[36]。
IR边界的扩展与收缩是导致叶绿体基因组的长度和结构存在差异的重要原因[37]。通过分析Hygrophila megalantha和Hygrophila sp.的叶绿体基因组中的IR/SC边界发现,LSC/IRb、IRb/SSC、SSC/IRa3个边界高度保守,分布的基因种类和大小较为相似;两者的主要差异集中于JLA边界:Hygrophila megalantha的trnH基因横跨JLA边界,而Hygrophila sp.的trnH基因位于LSC区域内。这些差异为Hygrophila megalantha物种鉴定提供了潜在序列资源。
基于大花水蓑衣和Hygrophila sp.的叶绿体基因组研究,可以初步了解两者叶绿体基因组的结构特征及系统发育关系。虽然对于Hygrophila sp.不能定种,但它是广天仙子在市场上的常见混伪品,后续研究可以基于大花水蓑衣、Hygrophila sp.和其他混伪品叶绿体基因组上的碱基序列差异开发分子标记用于中药材广天仙子的鉴别,也为进一步研究大花水蓑衣及属内物种的进化关系提供数据基础。
| [1] |
国家食品药品监督管理局. 关于颁布儿茶等43种进口药材质量标准的通知: 国食药监注[2004]144号[A/OL]. (2004-05-08)[2024-02-26]. https://www.nmpa.gov.cn/xxgk/fgwj/gzwj/gzwjyp/20040508010101798.html.
|
| [2] |
张有礼. 南天仙子外敷治疗急性乳腺炎50例[J]. 中国中西医结合杂志, 1992, 12(7): 42. |
| [3] |
曾小芳, 孙海玲, 方少意, 等. 应用中西医结合方法治疗大面积深度褥疮[J]. 现代护理, 2003, 9(5): 373. DOI:10.3760/cma.j.issn.1674-2907.2003.05.031 |
| [4] |
曾小芳, 陈淑贤, 许桂华, 等. 南天仙子外敷与皮维碘外敷治疗压疮的疗效比较[J]. 中国临床研究, 2013, 26(4): 405-406. |
| [5] |
曾小芳, 陈淑贤, 方少意, 等. 南天仙子外敷治疗化脓性感染创面的疗效观察[J]. 中国误诊学杂志, 2007, 7(16): 3714-3715. DOI:10.3969/j.issn.1009-6647.2007.16.011 |
| [6] |
朱文君. 中药南天仙子湿敷治疗甘露醇致静脉炎的效果观察[J]. 当代护士(学术版), 2011, 18(4): 139-140. DOI:10.3969/j.issn.1006-6411.2011.04.089 |
| [7] |
刘天付, 辛培尧. 叶绿体基因组及其在樟科植物系统发育研究中的应用[J]. 寒旱农业科学, 2024(3): 197-202. DOI:10.3969/j.issn.2097-2172.2024.03.001 |
| [8] |
郭华, 徐流巍, 邢志美, 等. 基于板蓝根及其混伪品叶绿体基因组的mini-barcode开发及其定性能力的研究[J]. 天津中医药, 2023, 40(5): 662-667. DOI:10.11656/j.issn.1672-1519.2023.05.21 |
| [9] |
JIN J J, YU W B, YANG J B, et al. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes[J]. Genome Biology, 2020, 21(1): 241. DOI:10.1186/s13059-020-02154-5 |
| [10] |
KEARSE M, MOIR R, WILSON A, et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data[J]. Bioinformatics, 2012, 28(12): 1647-1649. DOI:10.1093/bioinformatics/bts199 |
| [11] |
QU X J, MOORE M J, LI D Z, et al. PGA: A software package for rapid, accurate, and flexible batch annotation of plastomes[J]. Plant Methods, 2019, 15: 50. DOI:10.1186/s13007-019-0435-7 |
| [12] |
SHI L C, CHEN H M, JIANG M, et al. CPGAVAS2, an integrated plastome sequence annotator and analyzer[J]. Nucleic Acids Research, 2019, 47(W1): DOI: 10.1093.
|
| [13] |
TILLICH M, LEHWARK P, PELLIZZER T, et al. GeSeq-versatile and accurate annotation of organelle genomes[J]. Nucleic Acids Research, 2017, 45(W1): DOI: 10.1093.
|
| [14] |
CHAN P P, LOWE T M. tRNAscan-SE Searching for tRNA Genes in Genomic Sequences[J]. Methods in Molecular Biology, 2019, 1962: 1-14. |
| [15] |
LAGESEN K, HALLIN P, RØDLAND E A, et al. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes[J]. Nucleic Acids Research, 2007, 35(9): 3100-3108. DOI:10.1093/nar/gkm160 |
| [16] |
BENSON D A, CAVANAUGH M, CLARK K, et al. GenBank[J]. Nucleic Acids Research, 2013, 41: D36-42. DOI:10.1093/nar/gks1035 |
| [17] |
ZHENG S Y, POCZAI P, HYVÖNEN J, et al. Chloroplot: An online program for the versatile plotting of organelle genomes[J]. Frontiers in Genetics, 2020, 11: 576124. DOI:10.3389/fgene.2020.576124 |
| [18] |
FRAZER K A, LIOR P, ALEXANDER P, et al. VISTA: computational tools for comparative genomics[J]. Nucleic Acids Research, 2004, 32: W273-W279. DOI:10.1093/nar/gkh458 |
| [19] |
KUMAR S, STECHER G, LI M, et al. MEGA X: Molecular evolutionary genetics analysis across computing platforms[J]. Molecular Biology and Evolution, 2018, 35(6): 1547-1549. DOI:10.1093/molbev/msy096 |
| [20] |
SEBASTIAN B, THOMAS T, MÜNCH T, et al. MISA-web: a web server for microsatellite prediction[J]. Bioinformatics, 2017, 33(16): 2583-2585. DOI:10.1093/bioinformatics/btx198 |
| [21] |
KURTZ S, CHOUDHURI J V, OHLEBUSCH E, et al. REPuter: The manifold applications of repeat analysis on a genomic scale[J]. Nucleic Acids Research, 2001, 29(22): 4633-4642. DOI:10.1093/nar/29.22.4633 |
| [22] |
AMIRYOUSEFI A, HYVÖNEN J, POCZAI P. IRscope: An online program to visualize the junction sites of chloroplast genomes[J]. Bioinformatics, 2018, 34(17): 3030-3031. DOI:10.1093/bioinformatics/bty220 |
| [23] |
ZHANG D, GAO F L, JAKOVLIĆ I, et al. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies[J]. Molecular Ecology Resources, 2020, 20(1): 348-355. DOI:10.1111/1755-0998.13096 |
| [24] |
NGUYEN L T, SCHMIDT H A, VON HAESELER A, et al. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies[J]. Molecular Biology and Evolution, 2015, 32(1): 268-274. DOI:10.1093/molbev/msu300 |
| [25] |
SHARP P M, LI W H. The Codon Adaptation Index: A measure of directional synonymous Codon usage bias, and its potential applications[J]. Nucleic Acids Research, 1987, 15(3): 1281-1295. DOI:10.1093/nar/15.3.1281 |
| [26] |
张加强, 史小华, 刘慧春, 等. '京红久'忍冬叶绿体基因组微卫星序列特征分析[J]. 分子植物育种, 2023, 21(6): 1955-1966. |
| [27] |
HUANG H, SHI C, LIU Y, et al. Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: Genome structure and phylogenetic relationships[J]. BMC Evolutionary Biology, 2014, 14: 151. DOI:10.1186/1471-2148-14-151 |
| [28] |
段义忠, 张凯. 沙冬青属植物叶绿体基因组对比和系统发育分析[J]. 西北植物学报, 2020, 40(8): 1323-1332. |
| [29] |
HU J H, GUI S T, ZHU Z X, et al. Genome-wide identification of SSR and SNP markers based on whole-genome re-sequencing of a Thailand wild sacred Lotus(Nelumbo nucifera)[J]. PLoS One, 2015, 10(11): e0143765. DOI:10.1371/journal.pone.0143765 |
| [30] |
王化坤, 娄晓鸣, 章镇. 叶绿体微卫星在植物种质资源研究中的应用[J]. 分子植物育种, 2006, 4(S1): 92-98. |
| [31] |
王海阁, 许文, 张勋, 等. 花叶开唇兰与台湾银线兰的SSR标记技术鉴别[J]. 中草药, 2022, 53(3): 848-852. |
| [32] |
严卓彦, 夏瑛瑛, 崔洁, 等. 基于DNA条形码及SSR技术鉴定风藤和山蒟[J]. 现代中药研究与实践, 2023, 37(2): 23-27. |
| [33] |
林汇源, 何楚扬, 张启赢, 等. 植物基因组重复DNA序列与基因组大小[J/OL]. 分子植物育种, 1-14[2025-04-28]. http://kns.cnki.net/kcms/detail/46.1068.S.20230425.1450.019.html.
|
| [34] |
李新玉, 王希胤. 重复序列对植物基因组大小进化的影响[J]. 华北理工大学学报(自然科学版), 2021, 43(4): 98-107. DOI:10.3969/j.issn.2095-2716.2021.04.015 |
| [35] |
杨国锋, 苏昆龙, 赵怡然, 等. 蒺藜苜蓿叶绿体密码子偏好性分析[J]. 草业学报, 2015, 24(12): 171-179. DOI:10.11686/cyxb2015016 |
| [36] |
SUZUKI Y. Statistical methods for detecting natural selection from genomic data[J]. Genes & Genetic Systems, 2010, 85(6): 359-376. |
| [37] |
LI L, HU Y F, HE M, et al. Comparative chloroplast genomes: Insights into the evolution of the chloroplast genome of Camellia sinensis and the phylogeny of Camellia[J]. BMC Genomics, 2021, 22(1): 138. DOI:10.1186/s12864-021-07427-2 |