 2022, Vol. 41
2022, Vol. 41文章信息
- 王华玲, 邓霞, 任强, 周金辉
- WANG Hualing, DENG Xia, REN Qiang, ZHOU Jinhui
- HPLC-QAMS法同时定量分析归芪补血口服液中8种成分含量
- Simultaneous quantitative analysis of eight components in Guiqi Buxue Oral Liquid by HPLC-QAMS
- 天津中医药大学学报, 2022, 41(5): 630-637
- Journal of Tianjin University of Traditional Chinese Medicine, 2022, 41(5): 630-637
- http://dx.doi.org/10.11656/j.issn.1673-9043.2022.05.17
-
文章历史
收稿日期: 2022-04-27

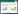




2. 济宁医学院药学院,日照 276800
2. College of Pharmacy, Jining Medical University, Rizhao 276800, China
归芪补血口服液由党参、黄芪、茯苓、丹参等12味中药材组方而成,临床上主要用于治疗气血不足、气虚血瘀之少气懒言、神疲乏力、心悸失眠、头晕目眩等病症。现代研究表明归芪补血口服液可显著升高失血性贫血小鼠血常规红细胞总数、血红蛋白含量、红细胞容积、网织红细胞计数、血小板总数和白细胞总数,具有良好的补血效果和免疫器官保护作用[1]。方中黄芪补气升阳、生津养血,当归补血活血,两者共为君药;党参、丹参活血祛瘀、养血生津,制何首乌、枸杞子、女贞子补肝肾、益精血,共为臣药;茯苓宁心安神,香菇、银耳补气生津、养阴理气,三七散瘀止血,共为佐药;甘草补脾益气、调和诸药,为其使药。诸药合用,共奏益气补血、活血祛瘀之功。现行的归芪补血口服液质量标准[2]和文献报道[3-4]仅对黄芪甲苷、阿魏酸等单体成分进行了定量控制,无法准确反映中成药复方制剂多成分-多靶点-多途径的整体观特点。通过建立多指标成分质量控制模式,不断摸索和完善中药及其制剂原药材源头控制和制剂生产过程控制,最终使产品质量和临床疗效达到稳定,已成为中药制剂质量控制的发展趋势。
本实验根据中医配伍组方理论,结合中药质量标志物的现代研究,以归芪补血口服液方中君药黄芪活性成分毛蕊异黄酮葡萄糖苷、芒柄花苷和9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷,臣药党参特征成分党参炔苷和紫丁香苷,佐药茯苓活性成分去氢土莫酸、去氢茯苓酸和茯苓酸为定量控制指标成分,采用高效液相色谱一测多评(HPLC-QAMS)法对所确定的8种定量控制指标成分含量同时进行测定,建立归芪补血口服液多指标成分质量控制方法,以期为指导药品生产企业和监管部门提升该制剂质量控制标准、逐步完善和稳定原药材来源与生产过程质量控制提供参考依据。
1 材料 1.1 实验仪器UltiMate 3000型高效液相色谱仪(美国赛默飞世尔科技公司)、Shimadzu LC-20A型高效液相色谱仪(日本岛津公司);Agilent Zorbax SB-C18色谱柱(250 mm×4.6 mm,5 μm)、Hedera C18色谱柱(250 mm×4.6 mm,5 μm)、Diamonsil C18色谱柱(250 mm×4.6 mm,5 μm);AB265-S型电子分析天平(瑞士梅特勒-托利多公司)。
1.2 药物及试剂归芪补血口服液(规格:每支10 mL,批号:20190905、20191206、20200305)来源于广西康晟制药有限责任公司。对照品紫丁香苷(批号:111574-201605,纯度95.2%)和毛蕊异黄酮葡萄糖苷(批号:111920-201907,纯度96.8%)来源于中国食品药品检定研究院;对照品党参炔苷(批号:PRF9121002,纯度99.9%)、芒柄花苷(批号:PRF8081121,纯度99.6%)和9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷(批号:PRF8101143,纯度99.1%)来源于成都普瑞法科技开发有限公司;对照品去氢土莫酸(批号:PS010100,纯度98.5%)、去氢茯苓酸(批号:PS010107,纯度91.2%)和茯苓酸(批号:PS010073,纯度99.7%)来源于成都普思生物科技股份有限公司;制备阴性供试品所用黄芪、当归、制何首乌、枸杞子、女贞子、党参、丹参、茯苓、香菇、银耳、三七和甘草均来源于济宁邦尔中药饮片有限公司,经检验符合各自质量标准项下规定;乙腈(色谱纯)来源于美国Fisher公司,其余试剂为分析纯。
2 方法与结果 2.1 混合对照品溶液的制备精密称取党参炔苷、紫丁香苷、毛蕊异黄酮葡萄糖苷、芒柄花苷、9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷、去氢土莫酸、去氢茯苓酸和茯苓酸对照品各适量,用70%甲醇制成混合对照品贮备液(折纯后党参炔苷0.382 mg/mL、紫丁香苷0.128 mg/mL、毛蕊异黄酮葡萄糖苷0.314 mg/mL、芒柄花苷0.196 mg/mL、9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷0.280 mg/mL、去氢土莫酸0.152 mg/mL、去氢茯苓酸0.116 mg/mL、茯苓酸0.232 mg/mL);精密吸取上述贮备液2.0 mL,用70%甲醇定容至50 mL,制成各成分质量浓度分别15.28、5.12、12.56、7.84、11.20、6.08、4.64和9.28 μg/mL的混合对照品溶液。
2.2 供试品溶液的制备精密吸取归芪补血口服液2.0 mL,用70%甲醇稀释至25 mL,摇匀后过滤,制得归芪补血口服液供试品溶液。按归芪补血口服液标准项下处方比例和制备过程分别制备缺少党参、缺少黄芪和缺少茯苓的阴性样品,按上述方法制备阴性供试品溶液。
2.3 色谱条件及专属性实验色谱柱为Agilent Zorbax SB-C18柱(250 mm×4.6 mm,5 μm),柱温为25 ℃。流动相为乙腈-0.05%磷酸水溶液,梯度洗脱(0~11 min,15.0%乙腈;11~18 min,15.0%→22.0%乙腈;18~29 min,22.0%→50.0%乙腈;29~47 min,50.0%→92.0%乙腈;47~60 min,92.0%→11.0%乙腈),流速为1.0 mL/min。检测波长分别为254 nm(0~29 min检测党参炔苷、紫丁香苷、毛蕊异黄酮葡萄糖苷、芒柄花苷和9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷)[5-12]和210 nm(29~60 min检测去氢土莫酸、去氢茯苓酸和茯苓酸)[13-15]。进样量为10 μL。精密吸取“2.1”“2.2”项下溶液各10 μL,注入高效液相色谱仪中,依法进样检测,结果党参炔苷、紫丁香苷、毛蕊异黄酮葡萄糖苷、芒柄花苷、9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷、去氢土莫酸、去氢茯苓酸和茯苓酸峰形对称,拖尾因子符合《中华人民共和国药典》规定;理论塔板数按各成分色谱峰计均≥3 500,且与相邻色谱峰能有效分离,分离度均 > 1.5;阴性供试品对归芪补血口服液中党参炔苷、紫丁香苷、毛蕊异黄酮葡萄糖苷、芒柄花苷、9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷、去氢土莫酸、去氢茯苓酸和茯苓酸的同时测定无干扰。见图 1。

|
| 注:A.混合对照品;B.归芪补血口服液;C.党参阴性供试品;D.黄芪阴性供试品;E.茯苓阴性供试品;1.党参炔苷;2.紫丁香苷;3.毛蕊异黄酮葡萄糖苷;4.芒柄花苷;5.9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷;6.去氢土莫酸;7.去氢茯苓酸;8.茯苓酸。 图 1 归芪补血口服液中党参炔苷、紫丁香苷、毛蕊异黄酮葡萄糖苷、芒柄花苷、9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷、去氢土莫酸、去氢茯苓酸和茯苓酸的高效液相色谱图 |
分别精密吸取“2.1”项下混合对照品贮备液各适量,用70%甲醇制成6个系列梯度浓度混合对照品溶液,各精密吸取10 μL,注入高效液相色谱仪中,依法进样测定党参炔苷、紫丁香苷、毛蕊异黄酮葡萄糖苷、芒柄花苷、9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷、去氢土莫酸、去氢茯苓酸和茯苓酸的峰面积,以峰面积对质量浓度进行线性回归,得到党参炔苷、紫丁香苷、毛蕊异黄酮葡萄糖苷、芒柄花苷、9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷、去氢土莫酸、去氢茯苓酸和茯苓酸的回归方程及线性范围。见表 1。
精密吸取批号为20190905的归芪补血口服液同一份供试品溶液10 μL,注入高效液相色谱仪中,依法进样测定党参炔苷、紫丁香苷、毛蕊异黄酮葡萄糖苷、芒柄花苷、9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷、去氢土莫酸、去氢茯苓酸和茯苓酸的峰面积,重复进样6次,测得8种成分峰面积的相对标准偏差(RSD)分别为1.10%、1.26%、0.91%、1.06%、0.87%、1.22%、1.31%和1.05%。
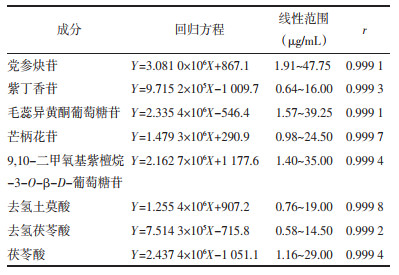
取批号为20190905的归芪补血口服液供试品,按“2.2”项方法平行制备6份供试品溶液,各精密吸取10 μL,注入高效液相色谱仪中,依法进样测定党参炔苷、紫丁香苷、毛蕊异黄酮葡萄糖苷、芒柄花苷、9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷、去氢土莫酸、去氢茯苓酸和茯苓酸的峰面积并计算各成分含量,结果8种成分含量的RSD分别为1.57%、0.85%、1.46%、0.62%、1.79%、1.83%、0.94%和1.72%。
取批号为20190905的归芪补血口服液同一份供试品溶液,于0、2、4、6、12、18、24 h各精密吸取10 μL,注入高效液相色谱仪中,依法进样测定党参炔苷、紫丁香苷、毛蕊异黄酮葡萄糖苷、芒柄花苷、9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷、去氢土莫酸、去氢茯苓酸和茯苓酸的峰面积。结果显示归芪补血口服液供试品溶液24 h内稳定,8种成分峰面积的RSD分别为1.06%、1.31%、1.05%、1.10%、0.79%、1.25%、1.36%和0.95%。
2.6 加样回收率实验取已知8种成分含量的批号为20190905的归芪补血口服液供试品9份,每份精密吸取1.0 mL,精密加入混合对照品溶液(党参炔苷0.184 mg/mL、紫丁香苷0.061 mg/mL、毛蕊异黄酮葡萄糖苷0.145 mg/mL、芒柄花苷0.087 mg/mL、9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷0.119 mg/mL、去氢土莫酸0.072 mg/mL、去氢茯苓酸0.048 mg/mL、茯苓酸0.093 mg/mL)0.8、1.0和1.2 mL各3份,对照品加入量约为原有量的80%、100%和120%,再按“2.2”项方法制备加样供试品溶液。精密吸取上述9种溶液各10 μL,注入高效液相色谱仪中,依法进样测定党参炔苷、紫丁香苷、毛蕊异黄酮葡萄糖苷、芒柄花苷、9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷、去氢土莫酸、去氢茯苓酸和茯苓酸的峰面积,计算得8种成分平均加样回收率及RSD分别为98.2%(0.97%)、97.0%(1.05%)、100.1%(0.76%)、98.7%(1.32%)、99.1%(1.43%)、97.2%(0.89%)、97.3%(1.38%)和98.4%(1.69%)。
2.7 相对校正因子的测定精密吸取“2.4”项下6个系列梯度浓度混合对照品溶液各10 μL,注入高效液相色谱仪中依法进样测定党参炔苷、紫丁香苷、毛蕊异黄酮葡萄糖苷、芒柄花苷、9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷、去氢土莫酸、去氢茯苓酸和茯苓酸的峰面积,以毛蕊异黄酮葡萄糖苷为内参物,按公式fk/s=fk/fs=(Xk×Ys)/(Xs×Yk)(式中X代表质量浓度,Y代表峰面积,s代表内参物,k代表其他成分)计算得党参炔苷、紫丁香苷、芒柄花苷、9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷、去氢土莫酸、去氢茯苓酸和茯苓酸的相对校正因子。见表 2。
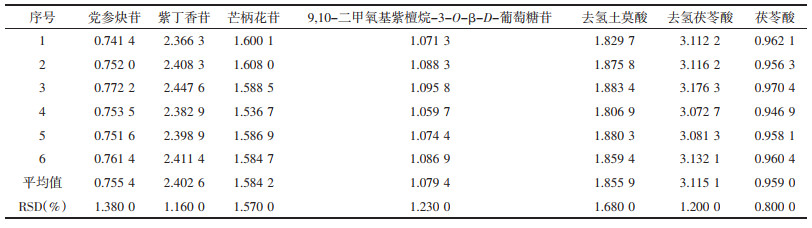
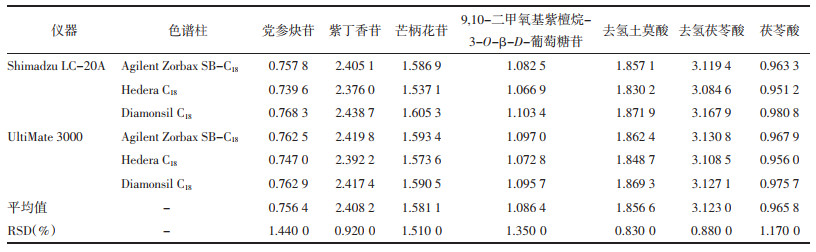
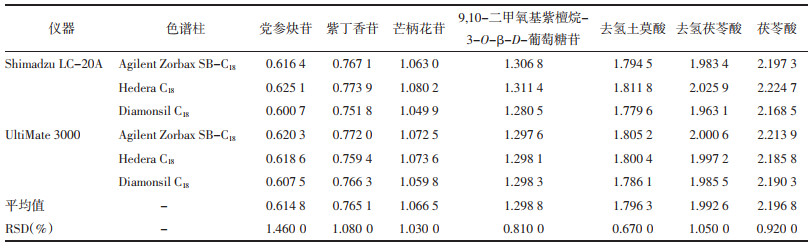
精密吸取“2.1”项下混合对照品溶液10 μL,注入高效液相色谱仪中依法进样,测定党参炔苷、紫丁香苷、毛蕊异黄酮葡萄糖苷、芒柄花苷、9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷、去氢土莫酸、去氢茯苓酸和茯苓酸的峰面积,对比考察Shimadzu LC-20A型、UltiMate 3000型高效液相色谱仪和Agilent Zorbax SB-C18、Hedera C18、Diamonsil C18色谱柱对相对校正因子的影响。见表 3。
精密吸取“2.1”项下混合对照品溶液10 μL,注入高效液相色谱仪中,依法进样测定党参炔苷、紫丁香苷、毛蕊异黄酮葡萄糖苷、芒柄花苷、9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷、去氢土莫酸、去氢茯苓酸和茯苓酸的峰面积,对比考察不同柱温(20、23、25、27、30 ℃)对相对校正因子的影响。见表 4。
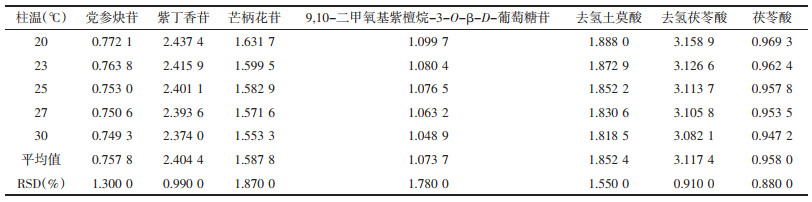
精密吸取“2.1”项下混合对照品溶液10 μL,注入高效液相色谱仪中,依法进样测定党参炔苷、紫丁香苷、毛蕊异黄酮葡萄糖苷、芒柄花苷、9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷、去氢土莫酸、去氢茯苓酸和茯苓酸的峰面积,对比考察不同流速(0.8、0.9、1.0、1.1、1.2 mL/min)对相对校正因子的影响。见表 5。
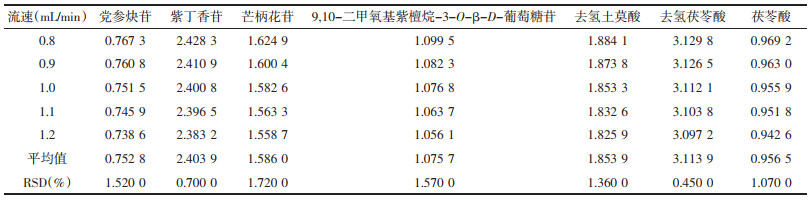
中成药复方制剂多指标成分色谱峰的准确定位,是确保所建立的HPLC-QAMS法具备准确性和可行性的基础。精密吸取“2.1”项下混合对照品溶液10 μL,注入高效液相色谱仪中,依法进样检测,记录党参炔苷、紫丁香苷、毛蕊异黄酮葡萄糖苷、芒柄花苷、9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷、去氢土莫酸、去氢茯苓酸和茯苓酸的保留时间,以内参物毛蕊异黄酮葡萄糖苷色谱峰为基准峰,采用相对保留时间值法在Shimadzu LC-20A型、UltiMate 3000型高效液相色谱仪和Agilent Zorbax SB-C18、Hedera C18、Diamonsil C18色谱柱条件下对色谱峰进行定位考察。见表 6。
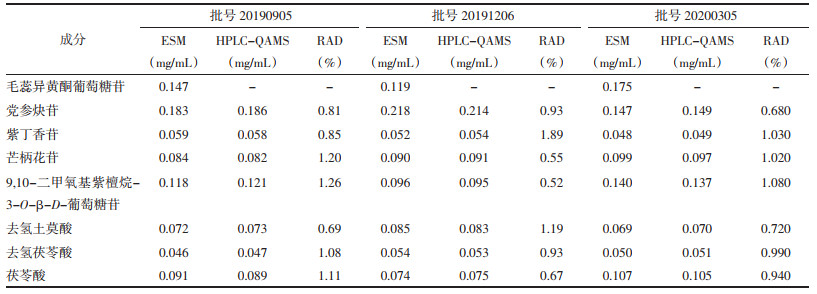
取3个批次的归芪补血口服液供试品适量,按“2.2”项方法平行制备3份归芪补血口服液供试品溶液,各精密吸取10 μL,注入高效液相色谱仪中,依法进样测定党参炔苷、紫丁香苷、毛蕊异黄酮葡萄糖苷、芒柄花苷、9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷、去氢土莫酸、去氢茯苓酸和茯苓酸的峰面积,采用外标法(ESM)和HPLC-QAMS法分别计算各成分含量。见表 7。
中成药复方制剂所含成分复杂,各成分极性均存在一定差异,流动性等度洗脱模式难以确保待测成分的有效分离,故本实验选用梯度洗脱模式对归芪补血口服液中多成分含量进行同时测定。考虑到甲醇在低波长处存在紫外吸收,影响色谱图基线平稳和检测结果的准确性,因此在实验过程中笔者参考相关文献[5-15],对比考察了乙腈-水[6-8]、乙腈-甲酸溶液[5, 9-10, 12]和乙腈-磷酸水溶液[13-14]等流动相系统,结果发现应用乙腈-水流动相体系检测时,成分芒柄花苷、9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷和去氢茯苓酸不能达到有效分离;应用乙腈-甲酸溶液流动相体系检测时,色谱图基线不平稳,直接影响去氢土莫酸、去氢茯苓酸和茯苓酸含量测定结果;乙腈-磷酸水溶液流动相体系检测效果较好。在此基础上对磷酸水溶液浓度和梯度洗脱有机相与水相的比例不断优化,最终确定乙腈-0.05%磷酸水溶液[14-15]为流动相进行梯度洗脱,同时检测归芪补血口服液中党参炔苷、紫丁香苷、毛蕊异黄酮葡萄糖苷、芒柄花苷、9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷、去氢土莫酸、去氢茯苓酸和茯苓酸的含量。
3.2 检测结果分析从表 7归芪补血口服液中党参炔苷、紫丁香苷、毛蕊异黄酮葡萄糖苷、芒柄花苷、9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷、去氢土莫酸、去氢茯苓酸和茯苓酸含量测定结果可以看出,HPLC-QAMS法计算结果与ESM实测值结果之间无明显差异(RAD < 2.0%),表明本研究建立的HPLC-QAMS法可用于归芪补血口服液中党参炔苷、紫丁香苷、毛蕊异黄酮葡萄糖苷、芒柄花苷、9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷、去氢土莫酸、去氢茯苓酸和茯苓酸含量的同时测定。同时表 7的含量测定结果也反映出8种目标化学成分含量均存在一定的批间差异,尤其毛蕊异黄酮葡萄糖苷、党参炔苷、9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷和茯苓酸较为明显,表明本研究所建立的多指标成分质量控制方法对稳定归芪补血口服液的产品质量具有重要意义。
归芪补血口服液现行质量控制标准采用蒸发光散射检测器对黄芪所含黄芪甲苷进行了定量控制,仅检索到对该制剂所含黄芪甲苷、阿魏酸等单体成分进行定量分析的文献报道。考虑到黄芪甲苷的化学结构中无强紫外吸收基团,仅在201 nm左右存在较弱的紫外末端吸收,同时君药黄芪所含毛蕊异黄酮葡萄糖苷、芒柄花苷、9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷等黄酮类具有补气固表调节机体免疫功能的作用[16],党参炔苷和紫丁香苷为臣药党参的主要活性成分,去氢土莫酸、去氢茯苓酸和茯苓酸为佐药茯苓的特征成分,故选取上述成分作为定量控制指标成分。本研究采用HPLC-QAMS法对归芪补血口服液中8种成分党参炔苷、紫丁香苷、毛蕊异黄酮葡萄糖苷、芒柄花苷、9, 10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷、去氢土莫酸、去氢茯苓酸和茯苓酸含量同时进行测定,首次建立了归芪补血口服液多指标成分质量控制模式,所建立的方法操作简便快速,结果准确,为归芪补血口服液现有质量标准提供了补充和完善,对全面提升归芪补血口服液整体控制方法、确保产品质量和临床疗效一致性、指导临床合理用药提供了参考数据。
| [1] |
林彩霞, 王捷, 马劼, 等. 归芪补血口服液对失血性贫血小鼠血象的影响[J]. 海峡药学, 2017, 29(12): 22-24. LIN C X, WANG J, MA J, et al. Effect of guiqi blood-enriching oral solution on hemorrhagic Anemia in mice[J]. Strait Pharmaceutical Journal, 2017, 29(12): 22-24. DOI:10.3969/j.issn.1006-3765.2017.12.006 |
| [2] |
国家药典委员会. 国家药品标准: WS-5803(B-0803)-2014Z[S]. 北京: 国家食品药品监督管理局, 2014. National Pharmacopoeia Commission. National drug standards: WS-5803(B-0803)-2014Z[S]. Beijing: State Food and Drug Administration, 2014. |
| [3] |
黄贵明. HPLC法测定归芪补血口服液中阿魏酸的含量[J]. 广西医科大学学报, 2008, 25(5): 768-770. HUANG G M. Determination of ferulic acid in Guiqi Buxue Oral Liquid by HPLC[J]. Journal of Guangxi Medical University, 2008, 25(5): 768-770. DOI:10.3969/j.issn.1005-930X.2008.05.050 |
| [4] |
陈荣, 黄梦娴. HPLC法测定归芪补血口服液中黄芪甲苷的含量[J]. 中国药师, 2009, 12(1): 77-79. CHEN R, HUANG M X. Determination the content of astagaloside in guiqibuxue oral liquid by HPLC[J]. China Pharmacist, 2009, 12(1): 77-79. DOI:10.3969/j.issn.1008-049X.2009.01.032 |
| [5] |
于小红, 赵嵘, 代云桃, 等. 党参标准汤剂质量评价的建立[J]. 中国实验方剂学杂志, 2017, 23(7): 24-29. YU X H, ZHAO R, DAI Y T, et al. Quality evaluation of standard decoction of Codonopsis Radix[J]. Chinese Journal of Experimental Traditional Medical Formulae, 2017, 23(7): 24-29. |
| [6] |
张平, 李冬华, 王晓琳, 等. 不同产地及加工方法对党参饮片质量的影响[J]. 西部中医药, 2019, 32(8): 20-24. ZHANG P, LI D H, WANG X L, et al. Effects of different processing methods and different producing areas on the quality of Dangshen decoction pieces[J]. Western Traditional Chinese Medicine, 2019, 32(8): 20-24. DOI:10.3969/j.issn.1004-6852.2019.08.006 |
| [7] |
王炯蓉, 许小红, 宁俊艳, 等. 甘肃省不同产地党参中党参炔苷的含量分析[J]. 中国卫生检验杂志, 2019, 29(23): 2821-2824. WANG J R, XU X H, NING J Y, et al. Analysis of lobetyolin content in Radix Codonopsis from different producing areas in Gansu Province[J]. Chinese Journal of Health Laboratory Technology, 2019, 29(23): 2821-2824. |
| [8] |
陈前锋, 邓小艳, 祝慧凤. HPLC法测定不同产地党参中党参炔苷和丁香苷的含量[J]. 食品工业科技, 2016, 37(6): 64-67. CHEN Q F, DENG X Y, ZHU H F. Determination of lobetyolin and syringin in Codonopsis radix from different places by HPLC[J]. Science and Technology of Food Industry, 2016, 37(6): 64-67. |
| [9] |
史鑫波, 唐志书, 刘妍如, 等. HPLC-UV-ELSD法同时测定黄芪中黄芪皂苷和黄酮类成分[J]. 天然产物研究与开发, 2019, 31(3): 434-440, 458. SHI X B, TANG Z S, LIU Y R, et al. Simultaneous determination of astragaloside and flavonoids components in astragali Radix using HPLC-UV-ELSD[J]. Natural Product Research and Development, 2019, 31(3): 434-440, 458. |
| [10] |
张妍, 董琳, 雍婧姣, 等. HPLC法同时测定黄芪药材中10种黄酮类成分的含量[J]. 中国药房, 2017, 28(21): 2970-2973. ZHANG Y, DONG L, YONG J J, et al. Simultaneous determination of 10 flavonoids in Astragalus membranaceus by HPLC[J]. China Pharmacy, 2017, 28(21): 2970-2973. DOI:10.6039/j.issn.1001-0408.2017.21.25 |
| [11] |
李婷婷, 舒志恒, 于良, 等. 不同产地黄芪HPLC/DAD指纹图谱及主要黄酮成分含量测定[J]. 中国医院药学杂志, 2015, 35(13): 1182-1187. LI T T, SHU Z H, YU L, et al. HPLC/DAD fingerprints and determination of main flavones in Radix Astragali from different origins[J]. Chinese Journal of Hospital Pharmacy, 2015, 35(13): 1182-1187. |
| [12] |
汤丹, 曹东敏, 谭兰芳, 等. 基于核壳色谱技术的黄芪中8种异黄酮活性成分的快速定量分析[J]. 中国中药杂志, 2019, 44(7): 1410-1415. TANG D, CAO D M, TAN L F, et al. Simultaneous determination of 8 bioactive isoflavonoids in Astragali Radix by UHPLC equipped with core-shell column[J]. China Journal of Chinese Materia Medica, 2019, 44(7): 1410-1415. |
| [13] |
许甜甜, 金传山, 吴德玲, 等. 茯苓不同药用部位化学成分分析[J]. 安徽中医学院学报, 2013, 32(1): 77-79. XU T T, JIN C S, WU D L, et al. Analysis of chemical constituents in different medicinal parts of Poria Cocos[J]. Journal of Anhui Traditional Chinese Medical College, 2013, 32(1): 77-79. |
| [14] |
张靓琦, 贾英, 罗洁, 等. 超高效液相色谱法同时测定茯苓中去氢土莫酸等6种活性成分的含量[J]. 中国药学杂志, 2012, 47(13): 1080-1083. ZHANG L Q, JIA Y, LUO J, et al. UPLC Determination of determine dehydrotumulosic acid etc. 6 active components in Poria[J]. Chinese Pharmaceutical Journal, 2012, 47(13): 1080-1083. |
| [15] |
刘校妃, 李健康, 唐怡, 等. 茯苓中去氢土莫酸和茯苓酸含量的高效液相色谱波长切换法同时测定[J]. 时珍国医国药, 2016, 27(3): 516-518. LIU X F, LI J K, TANG Y, et al. Determination of dehydrotumulosic acid and pachymic acid in Poria by wavelength-switching HPLC[J]. Lishizhen Medicine and Materia Medica Research, 2016, 27(3): 516-518. |
| [16] |
杨冰, 于桂红, 李明雨, 等. 基于"补气固表"探究黄芪黄酮组分抑制C57BL/6荷瘤小鼠肿瘤生长及免疫调节机制研究[J]. 中国中药杂志, 2019, 44(23): 5184-5190. YANG B, YU G H, LI M Y, et al. Mechanism of flavonoid components in Astragali Radix in inhibiting tumor growth and immunoregulation in C57BL/6 tumor bearing mice based on "invigorating qi for consolidation of exterior"[J]. China Journal of Chinese Materia Medica, 2019, 44(23): 5184-5190. |