 2024, Vol. 43
2024, Vol. 43文章信息
- 吴雪, 邹纯才, 鄢海燕, 孔玉卓, 黄越
- WU Xue, ZOU Chuncai, YAN Haiyan, KONG Yuzhuo, HUANG Yue
- 正交实验优化瓜蒌皮微丸的制备工艺及其质量控制研究
- Optimization of preparation process of Trichosanthis Pericarpium micro-pills by orthogonal test and quality control study
- 天津中医药大学学报, 2024, 43(10): 892-899
- Journal of Tianjin University of Traditional Chinese Medicine, 2024, 43(10): 892-899
- http://dx.doi.org/10.11656/j.issn.1673-9043.2024.10.05
-
文章历史
收稿日期: 2024-05-06






2. 皖南医学院, 芜湖 241002;
3. 芜湖市食品药品检验中心, 芜湖 241002
2. Wannan Medical College, Wuhu 241002, China;
3. Wuhu Food and Drug Inspection Center, Wuhu 241002, China
瓜蒌皮(Trichosanthis Pericarpiu)为葫芦科植物瓜蒌(Trichosanthes kirilowii Maxim.)或双边瓜蒌(Trichosanthes rosthornii Harms)的干燥成熟果皮,具有清热化痰、宽胸散结之功效[1],常用于心绞痛、心力衰竭等心血管领域[2-4]。瓜蒌皮注射液上市后疗效显著,被广泛应用于临床,但因中药注射液的临床使用合理性等安全性问题[5-6],拟对瓜蒌皮注射液进行剂型改进。为此,本研究在预实验基础上利用挤出滚圆法制备瓜蒌皮微丸[7-8],以圆整度、Hausner比值、脆碎度及得率为评价指标,在单因素实验的基础上采用正交实验优化[9-10],并对优化结果进行验证。为更好地控制其质量,本研究参照2020年版《中华人民共和国药典》丸剂的质量控制要求,通过外观描述,明确瓜蒌皮微丸的圆整度、含水量、粒径、溶出度等参数水平,利用化学鉴别法(颜色反应)、薄层色谱法(TLC)鉴别其中的核苷成分[11],采用高效液相色谱(HPLC)法对瓜蒌皮微丸中腺苷等5种核苷类成分进行定量分析[12-14],对瓜蒌皮微丸进行质量控制,以期为瓜蒌皮微丸的进一步开发提供技术支持。
1 仪器与材料 1.1 仪器LGJ-10冷冻干燥机(北京松源华兴科技发展有限公司);Heidolph-LR4010/4011旋转蒸发仪(德国海道尔夫);JW5挤出滚圆机(江苏常州市佳腾制粒干燥设备有限公司);HAPDCS-2B脆碎度检查仪(北京恒奥德仪器仪表有限公司);RC-6智能溶出度测试仪(上海精密仪器仪表有限公司);OLYMPUS-BX53显微镜(日本奥林巴斯公司);LC-20AP高效液相色谱仪(日本岛津公司);KNT-2-20科特宁纯水机(合肥科宁特水处理设备有限公司);AUW-220 D型电子天平(日本岛津公司)。
1.2 材料瓜蒌皮(批号:20171202,河北安国市御颜坊中药材有限公司,经皖南医学院药学院生药学教研室包淑云副教授鉴定为葫芦科植物瓜蒌的干燥成熟果皮);微晶纤维素(批号:20180916,郑州市博研生物科技有限公司);淀粉(批号:110706,安徽山河辅料股份有限公司);α-萘酚(批号:20220512,上海展云化工有限公司);尿嘧啶(批号:65012334,国药集团化学试剂有限公司);黄嘌呤(批号:65013983,国药集团化学试剂有限公司);鸟嘌呤核苷(简称“鸟苷”,批号:65008360,国药集团化学试剂有限公司);腺嘌呤核苷(简称“腺苷”,批号:N68291,山东西亚化学工业有限公司);β-胸苷(简称“胸苷”,批号:I 3650,山东西亚化学工业有限公司);其他试剂为分析纯。
2 方法与结果 2.1 瓜蒌皮微丸的制备工艺研究以瓜蒌皮为原料,采用水提醇沉法[15]制备瓜蒌皮冻干粉。取瓜蒌皮冻干粉及辅料,加润湿剂制备软材,利用挤出滚圆机制备瓜蒌皮微丸(简称微丸)。所得微丸经40 ℃电热鼓风干燥3 h,过筛(18~24目),备用。
2.1.1 单因素实验 2.1.1.1 润湿剂考察微丸软材中一般以一定比例的乙醇水溶液为润湿剂,水与乙醇的比例及用量会影响软材的制备及微丸的圆整度、机械强度、孔隙率、释放度[16]。根据预实验结果,微晶纤维素与淀粉按照2∶1的比例,载药量为40%,滚圆频率25 Hz,滚圆时间2 min,设计不同比例的乙醇水溶液为润湿剂,分别以无水乙醇溶液、70%乙醇水溶液、50%乙醇水溶液和水为润湿剂,按“2.1”项下的方法制得微丸。随着水与乙醇的比例中乙醇的占比减小,微丸表面更加光滑,收率越高,圆整度值越小,Hausner比值与脆碎度无明显变化。结合实际情况,选择水为润湿剂。
2.1.1.2 辅料中微晶纤维素与淀粉含量比例考察根据预实验结果,选择润湿剂为水,载药量为40%,滚圆频率25 Hz,滚圆时间2 min,设计微晶纤维素与淀粉的比例分别为4∶1、3∶1、2∶1、1∶1、1∶2,按照“2.1”项下的方法制得微丸。在一定范围内,辅料中微晶纤维素越多则微丸越圆,辅料中淀粉越多则微丸表面越光滑。随着微晶纤维素比例增大,收率先增大后不变,Hausner比值增大。微晶纤维素与淀粉的比例为2∶1时,脆碎度最小。结合实际情况,选择微晶纤维素与淀粉的比例为2∶1。
2.1.1.3 载药量考察中药微丸的有效成分含量低容易导致载药量小而服用剂量大,但载药量过大不易成丸。根据预实验结果,选择润湿剂为水,微晶纤维素与淀粉按照2∶1的比例,滚圆频率25 Hz,滚圆时间2 min,设计载药量分别为30%、40%、50%,按照“2.1”项下的方法制得微丸。随着载药量增大,微丸黏度增大,圆整度增大,挤出的长条不易折断,滚圆后收率低,Hausner比值变化小,脆碎度先减小后不变。结合实际情况,选择载药量为40%。
2.1.1.4 滚圆频率考察根据预实验结果,选择润湿剂为水,微晶纤维素与淀粉比例为2∶1,载药量为40%,滚圆时间2 min,设计滚圆频率分别为15、25、35 Hz,按照“2.1”项下的方法制得微丸。随着滚圆频率增大,收率先增加后降低,圆整度无明显变化,Hausner比值先减小后增大,脆碎度先减小后不变。结合实际情况,选择滚圆频率为25 Hz。
2.1.1.5 滚圆时间考察选择润湿剂为水,微晶纤维素与淀粉比例为2∶1,载药量为40%,滚圆频率为25 Hz,设计滚圆时间分别为1、2、3 min,按照“2.1”项下的方法制得微丸。随着滚圆时间增加,微丸的收率先增加后降低,圆整度增大,Hausner比值无明显变化,脆碎度增大。结合实际情况,选择滚圆时间为2 min。
2.1.2 正交实验设计及结果 2.1.2.1 正交实验设计选择微晶纤维素与淀粉的比例(A)、载药量(B)、滚圆频率(C)、滚圆时间(D)为考察因素,以收率、圆整度、Hausner比值、脆碎度的综合评分为指标,利用SPSS 23.0软件设计L9(34)正交实验优选出最佳制备工艺。见表 1。
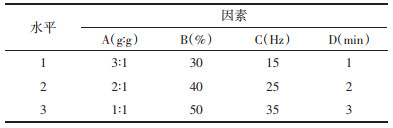
将测得的圆整度、Hausner比值、脆碎度和收率进行均值化的无量纲化法处理[17-18],得到均值化结果。根据实验实际情况最终确定圆整度、Hausner比值、脆碎度、收率的权重分别是0.1、0.2、0.3、0.4。计算微丸的收率、圆整度、Hausner比值、脆碎度均值化结果乘以各自权重的总和作为微丸制备工艺的综合评分(CII)。CII=(X1/X1)×0.1+(X2/X2)×0.2+(X3/X3)×0.3+(X4/X4)×0.4,其中X1、X2、X3、X4分别为圆整度、Hausner比值、脆碎度、收率的平均值。收率越高,综合评分越高。圆整度、Hausner比值、脆碎度越大,综合评分越低。正交实验结果见表 2。
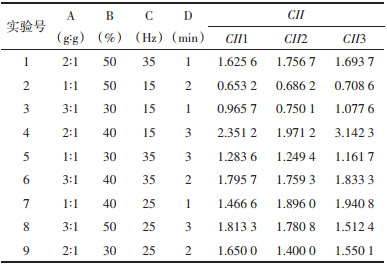
对表 2中的综合评分进行方差的单变量分析,以结果为因变量,以A、B、C、D为固定因子,选择相互交互模型,进行齐性检验,得到主体间效应检验表。结果见表 3。
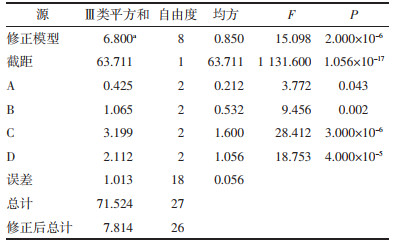
由表 3可知,4个影响因素的P值均小于0.01,表明4个因素对微丸的影响都有显著影响。将4个具有显著影响的因素依次输入“两两比较检验”的单因素ANOVA检验中,采用邓肯法进行计算,选择平均值图,结果见图 1。

|
| 图 1 各个因素的综合评分平均值 |
由图 1可知,最佳制备工艺为辅料(微晶纤维素与淀粉)的比例为2∶1,载药量40%,滚圆频率25 Hz,滚圆时间3 min。结合实际生产条件,确定最佳制剂工艺为:润湿剂为水,辅料(微晶纤维素与淀粉)比例为2∶1,载药量40%,滚圆频率25 Hz,滚圆时间3 min。
2.1.3 制剂工艺验证照按“2.1.2.2”项下的最佳制备工艺平行制备5批微丸,测得微丸的收率分别为91.8%、92.3%、93.0%、92.4%、93.8%,平均值为92.7%,相对标准偏差(RSD)值为0.83%;圆整度大小分别为20、18、20、19、19 °,平均值为19.2 °,RSD值为4.36%;Hausner比值分别为1.008、1.013、1.013、1.010、1.009,平均值为1.010 6,RSD值为0.23%;脆碎度分别为0.004 8、0.004 6、0.004 9、0.004 5、0.004 7,平均值为0.004 8,RSD值为3.36%。结果表明,微丸的制备工艺稳定可靠。
2.2 质量控制方法研究 2.2.1 性状分别取3批微丸(批号:2022091001、2022091002、2022091003),目视及显微镜下观察微丸的形状,明确颜色、气味。结果示:3批微丸肉眼可见呈黄色球状,无裂纹,无突起,焦糖气味;显微镜下观察,微丸呈圆形或椭圆形,无棒状、哑铃状微丸。见图 2。

|
| 图 2 瓜蒌皮微丸图 |
称取瓜蒌皮微丸1.25 g,瓜蒌皮冻干粉0.5 g,腺苷0.5 g,鸟苷0.5 g,空白辅料1.25 g,分别置于5个25 mL量瓶中,加水定容至刻度。取上述溶液各10 mL分别加至5个透析袋中并将透析袋置于装有25 mL水的试管内,静置24 h。取瓜蒌皮微丸透析液、瓜蒌皮冻干粉透析液、腺苷透析液、鸟苷透析液、空白辅料透析液各2 mL及纯水2 mL分别置于6个试管内,向6只试管中分别加入1 mL的5%α-萘酚溶液后,沿试管壁缓慢滴入浓硫酸20滴,静置3 min,观察试管内颜色变化。结果显示瓜蒌皮冻干粉、腺苷、鸟苷的颜色反应鉴别溶液中均有紫环生成,空白辅料和纯水没有出现紫环,说明瓜蒌皮微丸和瓜蒌皮冻干粉中存在与腺苷和鸟苷结构相同或相近的苷类物质,空白辅料和纯水对微丸的显色反应没有干扰。
2.2.2.2 TLC鉴别称取瓜蒌皮微丸(批号:2022091002)20 g,加水40 mL,搅匀,加入等量的乙酸乙酯萃取3次。取水层加入等量的正丁醇萃取3次,合并正丁醇层,减压回收正丁醇至一定体积后转移至5 mL量瓶内并用正丁醇定容至刻度得到瓜蒌皮微丸溶液。依法制得瓜蒌皮冻干粉溶液、空白辅料溶液。称取0.02 g腺苷,加正丁醇超声溶解,置于10 mL量瓶,加正丁醇定容至刻度制得对照品溶液。用毛细管分别蘸取瓜蒌皮微丸溶液、瓜蒌皮冻干粉溶液、对照品溶液、空白辅料溶液各10 μL,分别点于同一硅胶板(GF254薄层板110 ℃活化30 min),在展开剂(正丁醇∶乙酸乙酯∶水=5∶1∶5)为上层∶异丙醇∶浓氨水=10∶1∶0.1中展开8 cm后取出薄层板,晾干,置于紫外暗箱观察仪中,于254 nm的紫外灯下进行观察。见图 3。

|
| 注:1,瓜蒌皮微丸;2,瓜蒌皮冻干粉;3,腺苷;4,空白辅料。 图 3 瓜蒌皮微丸的薄层色谱图 |
由图 3可知,瓜蒌皮微丸和瓜蒌皮冻干粉与腺苷对照品在同一位置存在相同斑点,说明瓜蒌皮微丸中含有腺苷类成分,而空白辅料相应位置则没有斑点,说明辅料对薄层显色没有干扰。
2.2.3 检查 2.2.3.1 圆整度分别取5批瓜蒌皮微丸适量(批号:2022091001、2022091002、2022091003、2022091004、2022091005),将微丸置于光滑平板上,抬起平板一端使微丸出现滚动,记录出现滚动时的倾斜度,倾斜度越小说明微丸的圆整度越好。参照文献要求圆整度不得>30 °[19]。结果见表 4。
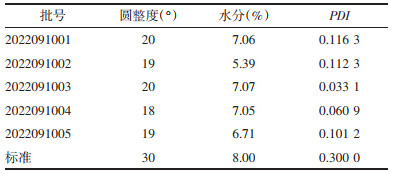
根据2020年版《中华人民共和国药典》水分测定法中的第二法(烘干法),分别取5批瓜蒌皮微丸各2.5 g(批号:2022091001、2022091002、2022091003、2022091004、2022091005),精密称定,平铺于扁形称量瓶中,置于105 ℃烘箱干燥5 h,通过烘干前后的质量差计算瓜蒌皮微丸的含水量。要求含水量≤8%。结果见表 4。
2.2.3.3 粒径取5批瓜蒌皮微丸适量(批号:2022091001、2022091002、2022091003、2022091004、2022091005),每批随机抽取10粒放置于光学显微镜下,计算微丸粒径的多分散指数(PDI)[20]。PDI=SD/D(SD代表标准偏差,D代表微丸的平均粒径)。当PDI<0.300 0时,表明微丸粒径均匀。结果见表 4。
2.2.3.4 溶出度1)专属性考察。对空白辅料、腺苷对照品、瓜蒌皮微丸于200~800 nm全波长扫描,结果显示空白辅料无吸收,腺苷和瓜蒌皮微丸在259 nm处吸收较强,表明空白辅料对瓜蒌皮微丸的核苷测定不会产生影响。
2)建立工作曲线。取腺苷对照品5 mg,精密称定,置于50 mL量瓶中,加水定容至刻度制得浓度为0.100 3 mg/mL的腺苷对照品溶液,备用。分别精密量取腺苷对照品溶液0.2、0.4、0.8、1.6、3.2 mL置于10 mL量瓶中,加水定容至刻度,于259 nm处分别测定其吸光度。以吸光度值A为纵坐标,以腺苷浓度c(mg/mL)为横坐标,建立回归方程:A=53.93c+0.001 7,r=0.999 5。结果表明腺苷在0.002~0.032 mg/mL线性关系良好。
3)总核苷含量测定。取瓜蒌皮微丸适量置于研钵中,研成细粉。取细粉2.001 7 g,精密称定,加至50 mL 0.1 mol/L盐酸溶液中,于室温搅拌30 min,用1%氢氧化钠溶液调至pH=7,离心10 min(转速5 000 r/min,离心半径10 cm)取上清液,并用0.1 mol/L盐酸溶液加至50 mL量瓶定容,0.45 μm滤膜过滤。取2 mL续滤液置于50 mL量瓶中,加水定容至刻度。于波长259 nm处测定瓜蒌皮微丸总核苷的吸光度值并计算得到总核苷浓度为0.222 6 mg/mL,即1 g瓜蒌皮微丸含总核苷质量为11.132 5 mg。
4)累积释放度测定。按照2020年版《中华人民共和国药典》第二法“浆法”测定瓜蒌皮微丸中总核苷的溶出度。取5批瓜蒌皮微丸各10 g(批号:2022091001、2022091002、2022091003、2022091004、2022091005),精密称定,置于装有900 mL 0.1 mol/L盐酸溶液的溶出杯中(满足漏漕条件),温度为(37±0.5)℃,分别于5、10、30、60、120 min自溶出杯取10 mL溶液,并补充等量同温的10 mL 0.1 mol/L盐酸溶液。按照“3)总核苷含量测定”方法测得各个时间点总核苷含量,计算瓜蒌皮微丸中总核苷的累积释放度,绘制时间-溶出度曲线。见图 4。

|
| 图 4 瓜蒌皮微丸总核苷累积释放度 |
由图 4可知,瓜蒌皮微丸中的总核苷类在5 min时累积释放度达到90%,10 min时累积释放度达到95%,60 min时累积释放度达到100%。说明微丸可以迅速释放药物,且能够释放完全。
2.2.4 微丸中5种核苷含量测定 2.2.4.1 溶液制备取瓜蒌皮微丸的研磨细粉2.0 g,精密称定,加水定容至10 mL,室温下充分搅拌,离心5 min(转速5 000 r/min,离心半径10 cm),取上清液,用0.45 μm滤膜过滤,续滤液备用。取续滤液2.5 mL置于5 mL量瓶中,加水定容至刻度,制成瓜蒌皮微丸供试品溶液。同法制备空白辅料溶液。分别取尿嘧啶0.01 g,腺苷0.01 g,鸟苷0.01 g,胸苷0.01 g,精密称定,置于10 mL量瓶中,加水定容至刻度,备用。取黄嘌呤0.05 g置于50 mL量瓶中,加水定容,备用。取1 mg/mL胸苷溶液1 mL,加水定容至10 mL,备用。取尿嘧啶(1 mg/mL)0.8 mL、黄嘌呤(0.1 mg/mL)2 mL、腺苷(1 mg/mL)0.8 mL、鸟苷(1 mg/mL)1.6 mL、胸苷(0.1 mg/mL)0.8 mL,置于100 mL量瓶中,加水至刻度,制成对照品混合溶液。
2.2.4.2 色谱条件色谱柱:YMC-Pack ODS-A(4.6 mm×250 mm,5 μm),流动相:甲醇(A)-0.1%磷酸溶液(B),0~10 min:0.1%A,10~30 min:0.1%A~3.5%A,30~50 min:3.5%A~12.0%A,50~85 min:12.0%A,流速:1 mL/min,柱温:25 ℃,检测波长:259 nm,进样量:20 μL。
2.2.4.3 建立标准曲线取“2.2.4.1”项下的尿嘧啶(1 mg/mL)1.6 mL、黄嘌呤(0.1 mg/mL)32 mL、腺苷(1 mg/mL)3.2 mL、鸟苷(1 mg/mL)3.2 mL、胸苷(0.1 mg/mL)3.2 mL置于100 mL量瓶中混均,加水定容作为混合对照品母液。将对照品母液分别稀释2、4、8、16倍。各对照品的浓度区间:尿嘧啶1~16 μg/mL、黄嘌呤1~16 μg/mL、腺苷1~16 μg/mL、鸟苷2~32 μg/mL、胸苷0.1~1.6 μg/mL。按照“2.2.4.2”项下色谱条件进样。以浓度c(μg/mL)为横坐标,峰面积y(mV×min)为纵坐标,分别建立尿嘧啶、黄嘌呤、腺苷、鸟苷、胸苷的标准曲线,得到尿嘧啶、黄嘌呤、腺苷、鸟苷、胸苷5种核苷的线性回归方程、相关系数(r)及线性范围。见表 5。
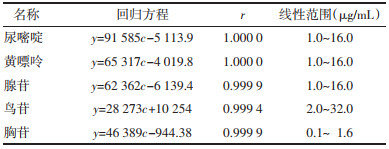
1)专属性考察。按照“2.2.4.1”项下的方法制成供试品溶液、对照品混合溶液、空白辅料溶液,按照“2.2.4.2”项下的色谱条件进样,得到瓜蒌皮微丸溶液、对照品混合溶液、空白辅料溶液的HPLC图。由图 5可知空白辅料对微丸没有影响。

|
| 注:A,瓜蒌皮微丸供试品溶液;B,混合对照品溶液;C,空白辅料溶液。1,尿嘧啶;2,黄嘌呤;3,腺苷;4,鸟苷;5,胸苷。 图 5 瓜蒌皮微丸供试品溶液、混合对照品溶液、空白辅料溶液的HPLC图 |
2)精密度考察。取瓜蒌皮微丸适量(批号:2002091002),按照“2.2.4.1”项下方法制成供试品溶液,按照“2.2.4.2”项下的色谱条件连续进样5次,计算5种核苷峰面积的RSD分别为0.12%、2.30%、0.15%、0.25%、2.06%,表明该方法精密度良好。
3)稳定性考察。取瓜蒌皮微丸适量(批号:2002091002),按照2.2.4.1”项下方法制成供试品溶液,按照“2.2.4.2”项下的色谱条件分别在0、60、120、180、240、300 min进样,计算各个时间点下5种核苷峰面积的RSD分别为0.27%、0.90%、0.04%、1.89%、2.80%,表明该方法稳定性良好。
4)重复性考察。分别取5批瓜蒌皮微丸适量(批号:2022091001、2022091002、2022091003、2022091004、2022091005),按照“2.2.4.1”项下方法制成供试品溶液,按照“2.2.4.2”项下的色谱条件进样,计算5批微丸的5种核苷峰面积的RSD分别为0.21%、1.35%、0.18%、2.73%、3.24%,表明该方法重复性良好。
5)加样回收率考察。取瓜蒌皮微丸适量(批号:2002091002),按照“2.2.4.1”项下方法制备9份供试品溶液,分为3组,依次向3组中分别加入所取供试品溶液中5种核苷已知质量的80%、100%、120%的各对照品溶液,按照“2.2.4.2”项下的色谱条件进样,9份供试品溶液中尿嘧啶、黄嘌呤、腺苷、鸟苷、胸苷5种核苷的平均加样回收率分别为98.34%、100.49%、99.47%、101.19%、98.82%,RSD分别为1.78%、1.81%、1.47%、1.23%、1.93%,表明该方法准确度良好。
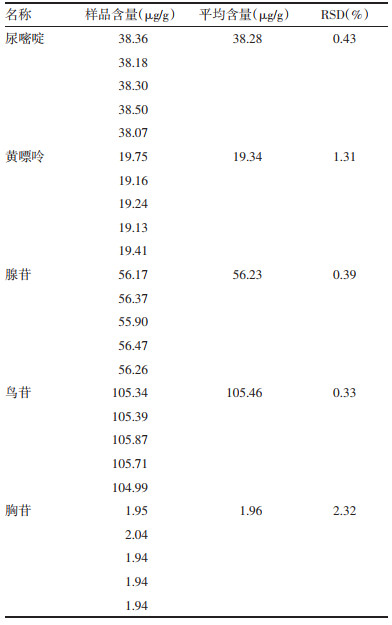
2.2.4.5 含量测定取5批瓜蒌皮微丸各2.0 g(批号:2022091001、2022091002、2022091003、2022091004、2022091005),按照“2.2.4.1”项下方法制备瓜蒌皮微丸供试品溶液,按照“2.2.4.2”项下的色谱条件进样,计算5种核苷含量。见表 6。
按照“2.2.4.1”项下方法取10批瓜蒌皮微丸适量(批号:2022091001、2022091002、2022091003、2022091004、2022091005、2022091006、2022091007、2022091008、2022091009、2022091010),制备10批瓜蒌皮微丸供试品溶液,按照“2.2.4.2”项下的色谱条件进样,使用中药色谱指纹图谱相似度评价系统软件(2012版)进行色谱图分析,以S1为参照物,进行Mark峰匹配,多点校正产生10批微丸的HPLC指纹图谱,见图 6(其中S1~S10为10批瓜蒌皮微丸色谱图,R为对照色谱图)。计算得到10批微丸相似度结果的平均值为0.998 7,RSD为0.067%,表明不同批次的瓜蒌皮微丸整体质量接近。

|
| 图 6 10批瓜蒌皮微丸的HPLC指纹图谱 |
本实验在制备瓜蒌皮微丸工艺优化过程中,除了以收率、圆整度、脆碎度为评价指标,还引入了Hausner比值以考察微丸的粉体流动性和填充性,有利于后期将微丸装入胶囊便于服用。实验中采用正交法优化微丸制备工艺。通常情况下,正交4个因素、3个水平实验处理过程中将影响因素较小的一组作为空白对照。本实验未设计空白因素作为对照,所以通过重复实验统计实验误差[21],即每个条件至少重复3次实验,得到9组共27次实验数据,结果采用SPSS软件处理后,通过均值图得到最优实验条件。
3.2 质量标准研究进行微丸粒径检查时,目视圆整的微丸在光学显微镜下并非显现为规则的圆形或椭圆,在测量微丸直径时考虑其各个方向粒径存在差异,可以将同一个微丸不同方向直径的平均直径作为该微丸的粒径,计算微丸的多分散指数。
瓜蒌皮微丸的颜色鉴别原理是苷类的Molish反应。苷水解生成的戊糖在浓硫酸条件下与α-萘酚发生紫环反应,微丸的辅料淀粉在浓硫酸条件下水解生成的葡萄糖也与α-萘酚发生紫环反应,因此使用透析袋过滤大分子,以避免淀粉对颜色鉴别造成的影响。在相同条件下,水、空白辅料、瓜蒌皮微丸、瓜蒌皮冻干粉、腺苷、鸟苷的透析液中,水和空白辅料都没有出现紫环反应,瓜蒌皮微丸、瓜蒌皮冻干粉、腺苷、鸟苷皆可见紫环反应,说明瓜蒌皮微丸与瓜蒌皮冻干粉中含有苷类成分。
参考国家食品药品监督管理局药品审评中心发布的《中药新药质量标准研究技术指导原则(试行)》,根据临床用药需求及剂型特征制定相应的指标标准。文献研究表明,核苷类成分是瓜蒌皮的抗血栓活性成分之一[22-24],因此本实验以核苷为对照品对瓜蒌皮微丸进行定性及定量研究,然而仅凭核苷这一类化学物质来制定瓜蒌皮微丸的质量标准存在一定局限性,后期将通过药效学实验及网络药理学研究进一步探索瓜蒌皮微丸的药效作用机制,完善瓜蒌皮微丸的质量标准。
| [1] |
国家药典委员会. 中华人民共和国药典: 一部[M]. 北京: 中国医药科技出版社, 2020: 116.
|
| [2] |
景贤, 赵映璇. 瓜蒌皮注射液联合磷酸肌酸治疗冠心病稳定型心绞痛的临床研究[J]. 现代药物与临床, 2022, 37(9): 2021-2025. |
| [3] |
代铁良. 瓜蒌皮注射液联合曲美他嗪在稳定型心绞痛治疗中的效果[J]. 实用中西医结合临床, 2021, 21(18): 62-63. |
| [4] |
冯祝婷, 钟金萍, 朱艳, 等. 瓜蒌皮注射液的研究概况[J]. 海峡药学, 2021, 33(10): 10-13. DOI:10.3969/j.issn.1006-3765.2021.10.004 |
| [5] |
程洁. 活血化瘀类中药注射液不良反应发生的影响因素[J]. 中国民康医学, 2022, 34(3): 109-111. DOI:10.3969/j.issn.1672-0369.2022.03.035 |
| [6] |
朱熠, 王双梅. 中药注射液高不良反应发生率问题探讨[J]. 北方药学, 2018, 15(8): 160-161. DOI:10.3969/j.issn.1672-8351.2018.08.134 |
| [7] |
李磊, 花丽, 王文龙. 挤出滚圆法制备淀粉微丸工艺研究[J]. 化工与医药工程, 2019, 40(3): 30-34. |
| [8] |
刘毅, 陈慧芳, 祝燕平, 等. 正交实验优选挤出滚圆法制备灯盏花素缓释微丸[J]. 牡丹江医学院学报, 2016, 37(5): 51-53. |
| [9] |
江蕾蕾, 王秀敏, 江昌照, 等. 大黄酸结肠靶向微丸包衣处方的筛选及其释药性能评价[J]. 中成药, 2022, 44(4): 1247-1250. DOI:10.3969/j.issn.1001-1528.2022.04.037 |
| [10] |
张雪, 胡志强, 李云琪, 等. 基于D-最优混料试验设计的挤出搓圆法制备丸剂的复合辅料研究[J]. 上海中医药大学学报, 2022, 36(1): 33-41. |
| [11] |
邹妍, 鄢海燕, 史玉婷, 等. 瓜蒌薤白滴丸的质量控制[J]. 中国医院药学杂志, 2014, 34(20): 1740-1743. |
| [12] |
曾棋平, 曹毅祥, 黄丽珊, 等. 复方首乌藤合剂的薄层鉴别和总黄酮的含量测定[J]. 药学实践杂志, 2016, 34(5): 424-427. |
| [13] |
毕继全, 王希通, 陈林伟, 等. 萌芽黑青稞喷干粉的主要成分测定及HPLC指纹图谱的建立[J]. 中国食品学报, 2022, 22(9): 293-303. |
| [14] |
张翠翠, 艾雯, 郭瑞齐, 等. 基于HPLC测定不同产地西洋参中12种人参皂苷含量及指纹图谱研究[J]. 辽宁中医杂志, 2022, 49(9): 144-147, 223. |
| [15] |
吴雪, 邹纯才, 鄢海燕, 等. Box-Behnken响应面法优化瓜蒌皮中总核苷和总氨基酸的提取工艺[J]. 右江民族医学院学报, 2022, 44(2): 211-217, 233. DOI:10.3969/j.issn.1001-5817.2022.02.014 |
| [16] |
黄洋, 贾晓斌, 陈斌. 水/乙醇在挤出滚圆法制备微丸中的作用与影响[J]. 中草药, 2010, 41(5): 845-848. |
| [17] |
李兴奇, 高晓红. 服从不同分布数据的无量纲化方法研究[J]. 统计与决策, 2022, 38(10): 31-36. |
| [18] |
邹纯才, 洪国君, 鄢海燕. 不同数据标准化方法对瓜蒌滴丸抗凝血作用谱效关系的影响[J]. 中国中药杂志, 2018, 43(9): 1864-1870. |
| [19] |
卢道元. 磷酸川芎嗪缓释微丸制备及质量评价[D]. 南昌: 江西中医药大学, 2021.
|
| [20] |
DANAEI M, DEHGHANKHOLD M, ATAEI S, et al. Impact of particle size and polydispersity index on the clinical applications of lipidic nanocarrier systems[J]. Pharmaceutics, 2018, 10(2): 57-68. DOI:10.3390/pharmaceutics10020057 |
| [21] |
王政, 魏莉. 利用SPSS软件实现药学实验中正交设计的方差分析[J]. 数理医药学杂志, 2014, 27(1): 99-102. |
| [22] |
刘岱琳, 曲戈霞, 王乃利, 等. 瓜蒌的抗血小板聚集活性成分研究[J]. 中草药, 2004, 35(12): 1334-1336. DOI:10.3321/j.issn:0253-2670.2004.12.005 |
| [23] |
邹纯才, 鄢海燕, 王莉丽, 等. 瓜蒌提取物抗血栓药效成分群的筛选与验证[J]. 药学学报, 2019, 54(3): 502-509. |
| [24] |
ASLAM M, SEDDING D, KOSHTY A, et al. Nucleoside triphosphates inhibit ADP, collagen, and epinephrine-induced platelet aggregation: role of P2Y1 and P2Y12 receptors[J]. Thrombosis Research, 2013, 132(5): 548-557. DOI:10.1016/j.thromres.2013.08.021 |